
Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation

 , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients and Plasma Samples
2.2. Stable Isotope Labeling of Protein Samples with 10 Plex Tandem Mass Tags
2.3. Nano-LC-MS/MS
2.4. Data Analysis
2.5. Calculation of the Analytical Variability and Inter-Individual Biological Variability
2.5.1. Analytical Variation
2.5.2. Inter-Individual Biological Variation
2.5.3. Sample Size Calculation
3. Results
3.1. The Proteome Dataset
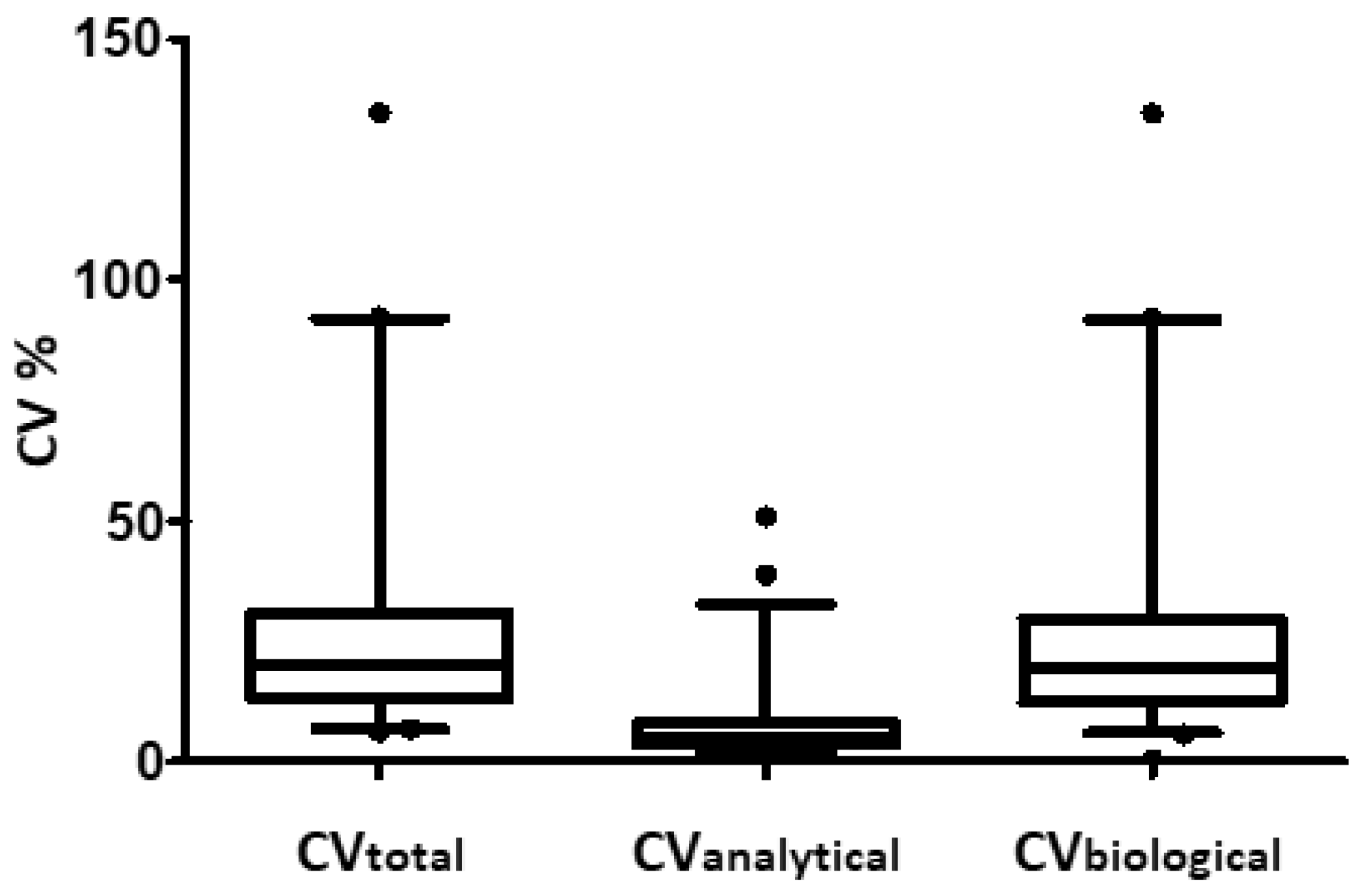
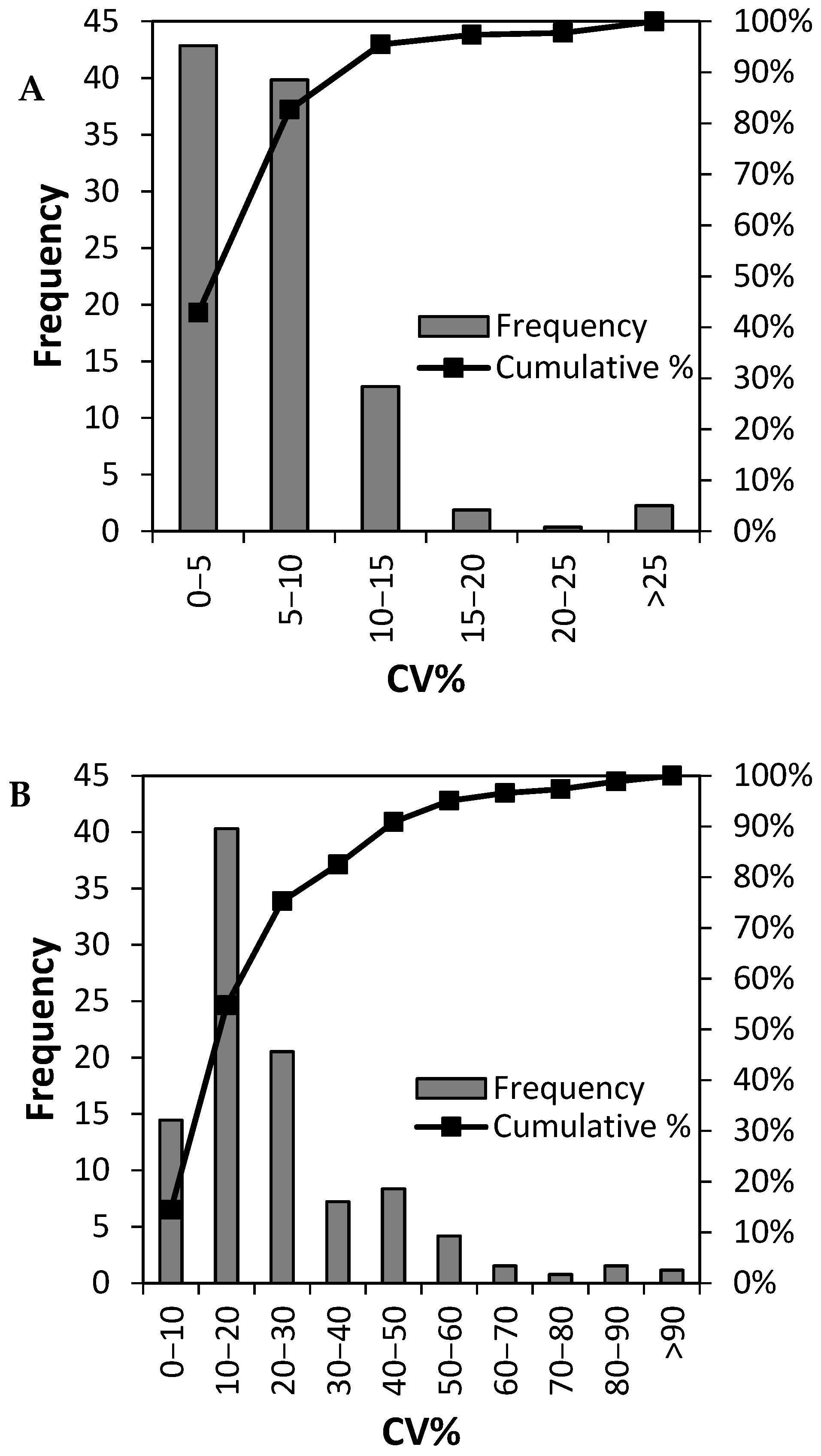
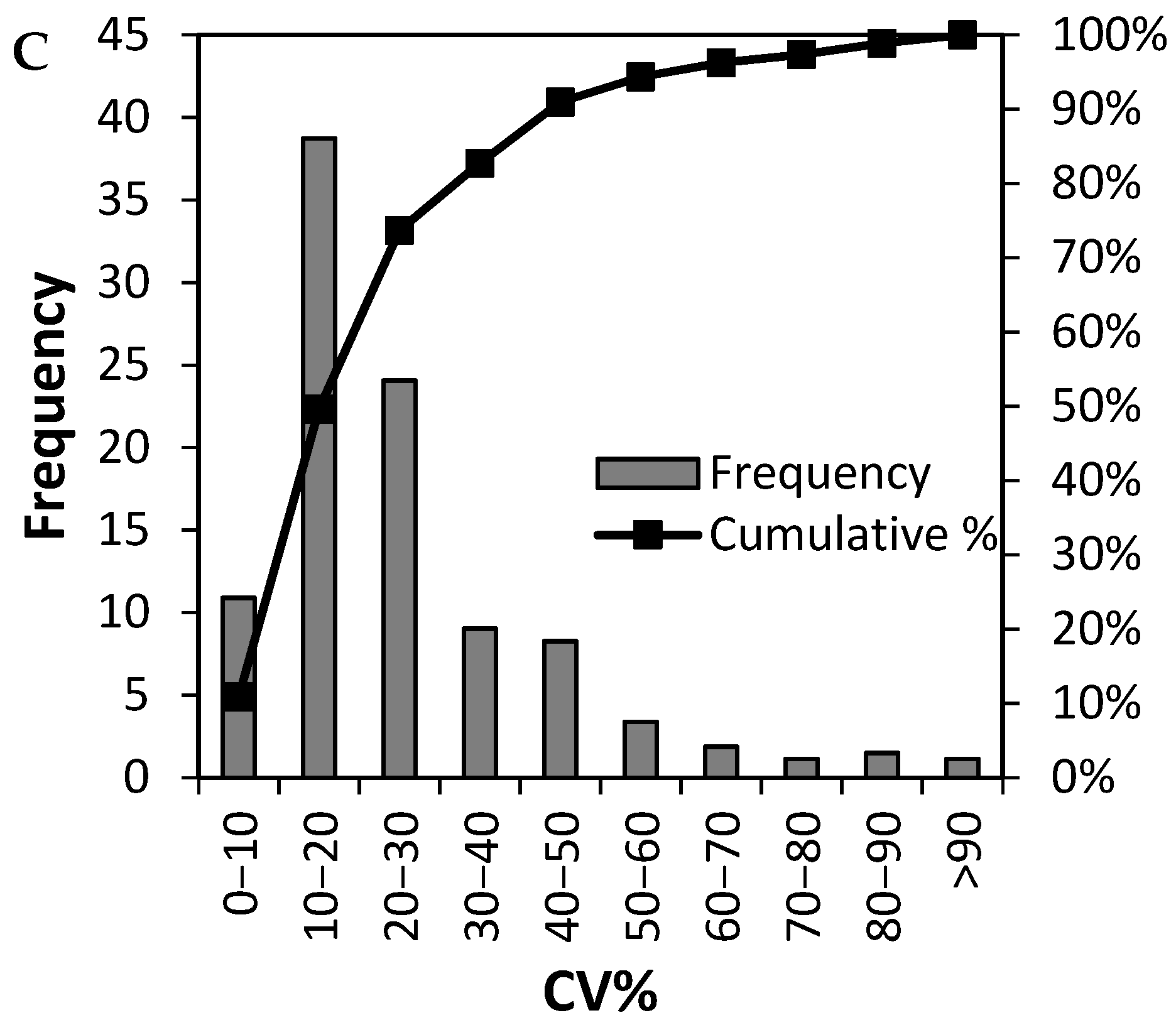
3.2. Analytical Precision of the LC-MSMS Method and Inter Individual Variation of 265 Plasma Proteins
3.3. Sample Size Determination
3.4. Can Biological Variation of Plasma Protein Be Determined by TMT-Based Relative Quantification
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aarsand, A.K.; Fernandez-Calle, P.; Webster, C.; Coskun, A.; Gonzales-Lao, E.; Diaz-Garzon, J.; Jonker, N.; Minchinela, J.; Simon, M.; Braga, F.; et al. The EFLM Biological Variation Database. Available online: https://biologicalvariation.eu/ (accessed on 25 March 2020).
- Wewer Albrechtsen, N.J.; Geyer, P.E.; Doll, S.; Treit, P.V.; Bojsen-Moller, K.N.; Martinussen, C.; Jørgensen, N.B.; Torekov, S.S.; Meier, F.; Niu, L.; et al. Plasma Proteome Profiling Reveals Dynamics of Inflammatory and Lipid Homeostasis Markers after Roux-En-Y Gastric Bypass Surgery. Cell Syst. 2018, 7, 601–612e3. [Google Scholar] [CrossRef] [Green Version]
- Beck, H.C.; Jensen, L.O.; Gils, C.; Ilondo, A.M.M.; Frydland, M.; Hassager, C.; Møller-Helgestad, O.K.; Møller, J.E.; Rasmussen, L.M. Proteomic Discovery and Validation of the Confounding Effect of Heparin Administration on the Analysis of Candidate Cardiovascular Biomarkers. Clin. Chem. 2018, 64, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Poljak, A.; Ali, S.A.; Zhong, L.; Raftery, M.J.; Sachdev, P. Extending the Depth of Human Plasma Proteome Coverage Using Simple Fractionation Techniques. J. Proteome Res. 2021, 20, 1261–1279. [Google Scholar] [CrossRef] [PubMed]
- Palstrom, N.B.; Rasmussen, L.M.; Beck, H.C. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. Int. J. Mol. Sci. 2020, 21, 5903. [Google Scholar] [CrossRef]
- Corzett, T.H.; Fodor, I.K.; Choi, M.W.; Walsworth, V.L.; Chromy, B.A.; Turteltaub, K.W.; McCutchen-Maloney, S.L. Statistical analysis of the experimental variation in the proteomic characterization of human plasma by two-dimensional difference gel electrophoresis. J. Proteome Res. 2006, 5, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, A.T.; Ilgisonis, E.V.; Moysa, A.A.; Tikhonova, O.V.; Zavialova, M.G.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.A.; Moshkovskii, S.A.; Markin, A.A. Targeted Quantitative Screening of Chromosome 18 Encoded Proteome in Plasma Samples of Astronaut Candidates. J. Proteome Res. 2016, 15, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Sundkvist, A.; Myte, R.; Boden, S.; Enroth, S.; Gyllensten, U.; Harlid, S.; van Guelpen, B. Targeted plasma proteomics identifies a novel, robust association between cornulin and Swedish moist snuff. Sci. Rep. 2018, 8, 2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshghi, A.; Pistawka, A.J.; Liu, J.; Chen, M.; Sinclair, N.J.T.; Hardie, D.B.; Elliott, M.; Chen, L.; Newman, R.; Mohammed, Y. Concentration Determination of >200 Proteins in Dried Blood Spots for Biomarker Discovery and Validation. Mol. Cell. Proteomics 2020, 19, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Perrin, R.J.; Payton, J.E.; Malone, J.P.; Gilmore, P.; Davis, A.E.; Xiong, C.; Fagan, A.M.; Townsend, R.R.; Holtzman, D.M. Quantitative label-free proteomics for discovery of biomarkers in cerebrospinal fluid: Assessment of technical and inter-individual variation. PLoS ONE 2013, 8, e64314. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, Y.; Zou, L.; Wang, D.; Shao, C.; Wang, Y.; Sun, W.; Zhang, L. A Proteomic Analysis of Individual and Gender Variations in Normal Human Urine and Cerebrospinal Fluid Using iTRAQ Quantification. PLoS ONE 2015, 10, e0133270. [Google Scholar] [CrossRef] [Green Version]
- Diederichsen, A.C.P.; Rasmussen, L.M.; Søgaard, R.; Lambrechtsen, J.; Steffensen, F.H.; Frost, L.; Egstrup, K.; Urbonaviciene, G.; Busk, M.; Olsen, M.H.; et al. The Danish Cardiovascular Screening Trial (DANCAVAS): Study protocol for a randomized controlled trial. Trials 2015, 16, 554. [Google Scholar] [CrossRef] [Green Version]
- Kadam, P.; Bhalerao, S. Sample size calculation. Int. J. Ayurveda Res. 2010, 1, 55–57. [Google Scholar] [PubMed] [Green Version]
- Motulsky, H. Intuitive Biostatistics, 4th ed.; Oxford University Press: Oxford, UK, 2018; p. 247. [Google Scholar]
- Carobene, A.; Aarsand, A.K.; Bartlett, W.A.; Coskun, A.; Diaz-Garzon, J.; Fernandez-Calle, P.; Guerra, E.; Jonker, N.; Locatelli, M.; Plebani, M.; et al. The European Biological Variation Study (EuBIVAS): A summary report. Clin. Chem. Lab. Med. 2021, 370. [Google Scholar] [CrossRef]
- Zhou, C.; Simpson, K.L.; Lancashire, L.; Walker, M.J.; Dawson, M.J.; Unwin, R.; Rembielak, A.; Price, P.; West, C.; Dive, C.; et al. Statistical Considerations of Optimal Study Design for Human Plasma Proteomics and Biomarker Discovery. J. Proteome Res. 2012, 11, 2103–2113. [Google Scholar] [CrossRef]
- Braga, F.; Panteghini, M. Biologic variability of C-reactive protein: Is the available information reliable? Clin. Chim. Acta 2012, 413, 1179–1183. [Google Scholar] [CrossRef]
- Clark, G.H.; Fraser, C.G. Biological variation of acute phase proteins. Ann. Clin. Biochem. 1993, 30 Pt 4, 373–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carobene, A.; Aarsand, A.K.; Guerra, E.; Bartlett, W.A.; Coskun, A.; Diaz-Garzon, J.; Fernandez-Calle, P.; Jonker, N.; Locatelli, M.; Sandberg, S. European Biological Variation Study (EuBIVAS): Within- and Between-Subject Biological Variation Data for 15 Frequently Measured Proteins. Clin. Chem. 2019, 65, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O., 3rd; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L. Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511. [Google Scholar]
- Ridker, P.M.; Morrow, D.A. C-reactive protein, inflammation, and coronary risk. Cardiol. Clin. 2003, 21, 315–325. [Google Scholar] [CrossRef]
- Zimmerman, L.J.; Li, M.; Yarbrough, W.G.; Slebos, R.J.C.; Liebler, D.C. Global Stability of Plasma Proteomes for Mass Spectrometry-Based Analyses. Mol. Cell. Proteomics 2012, 11, 014340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; The Consortium for Top Down Proteomics; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappireddi, N.; Martin, L.; Wuhr, M. A Review on Quantitative Multiplexed Proteomics. Chembiochem 2019, 20, 1210–1224. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Number of Patients (n) | 42 |
|---|---|
| Age range (mean) | 64–74 (68) |
| BMI range (mean) | 21.5–42.1 (28.2) |
| Male sex (%) | 41 (97.6) |
| CRP range (mg/L) (mean) | 0.6–98 (6.8) |
| LDL range (mmol/L) (mean) | 0.4–7.3 (4.6) |
| HDL range (mmol/L) (mean) | 0.8–2.6 (1.2) |
| Effect Size | Variance (Percentile) | ||||
|---|---|---|---|---|---|
| 70th | 75th | 80th | 85th | Maximum | |
| 1.1 | 131 (89–213) | 177 (120–288) | 233 (158–379) | 314 (213–511) | 2848 (1928–4630) |
| 1.2 | 33 (22–53) | 44 (30–72) | 58 (39–95) | 79 (53–128) | 712 (482–1158) |
| 1.5 | 5 (4–9) | 7 (5–12) | 9 (6–15) | 13 (9–20) | 114 (77–185) |
| 2.0 | 1 (1–2) | 2 (1–3) | 2 (2–4) | 3 (2–5) | 28 (19–46) |
| Uniprot Accession | Description | CVtotal % | CVanalytical % | CVbiological % | EFLM Biological Variation |
|---|---|---|---|---|---|
| P17936 | Insulin-like growth factor-binding protein 3 | 15.0 | 4.0 | 14.4 | 0.003 * |
| P02647 | Apolipoprotein A-I | 13.0 | 2.3 | 12.8 | 11.2 |
| P01034 | Cystatin-C | 16.3 | 6.6 | 14.9 | 12.1 |
| P05543 | Thyroxine-binding globulin | 9.4 | 3.3 | 8.8 | 12.6 * |
| P01024 | Complement C3 | 10.0 | 1.8 | 9.8 | 15.2 |
| P02766 | Transthyretin | 18.2 | 10.6 | 14.7 | 19.1 |
| P04114 | Apolipoprotein B-100 | 23.3 | 1.7 | 23.2 | 20.2 |
| P19652 | Alpha-1-acid glycoprotein 2 | 37.4 | 10.7 | 35.8 | 24.1 |
| P02763 | Alpha-1-acid glycoprotein 1 | 24.6 | 6.2 | 23.8 | 24.1 |
| P0C0L4 | Complement C4-A | 25.4 | 7.6 | 24.2 | 24.5 |
| P0C0L5 | Complement C4-B | 23.8 | 11.2 | 21.0 | 24.5 |
| P04278 | Sex hormone-binding globulin | 22.1 | 6.2 | 21.2 | 35.6 |
| P00738 | Haptoglobin | 38.3 | 2.7 | 38.2 | 39.0 |
| P02768 | Serum albumin | 7.1 | 1.9 | 6.8 | 5.1 |
| Q15848 | Adiponectin | 23.9 | 12.6 | 20.3 | 51.2 |
| P01009 | Alpha-1-antitrypsin | 13.7 | 2.8 | 13.4 | 10.5 |
| P02741 | C-reactive protein | 92.3 | 7.6 | 92.0 | 87.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, L.-A.C.; Palstrøm, N.B.; Diederichsen, A.; Lindholt, J.S.; Rasmussen, L.M.; Beck, H.C. Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation. Proteomes 2021, 9, 47. https://doi.org/10.3390/proteomes9040047
Andersen L-AC, Palstrøm NB, Diederichsen A, Lindholt JS, Rasmussen LM, Beck HC. Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation. Proteomes. 2021; 9(4):47. https://doi.org/10.3390/proteomes9040047
Chicago/Turabian StyleAndersen, Lou-Ann C., Nicolai Bjødstrup Palstrøm, Axel Diederichsen, Jes Sanddal Lindholt, Lars Melholt Rasmussen, and Hans Christian Beck. 2021. "Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation" Proteomes 9, no. 4: 47. https://doi.org/10.3390/proteomes9040047
APA StyleAndersen, L.-A. C., Palstrøm, N. B., Diederichsen, A., Lindholt, J. S., Rasmussen, L. M., & Beck, H. C. (2021). Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation. Proteomes, 9(4), 47. https://doi.org/10.3390/proteomes9040047