
Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Common Themes
3. Software Architecture and Data Formats
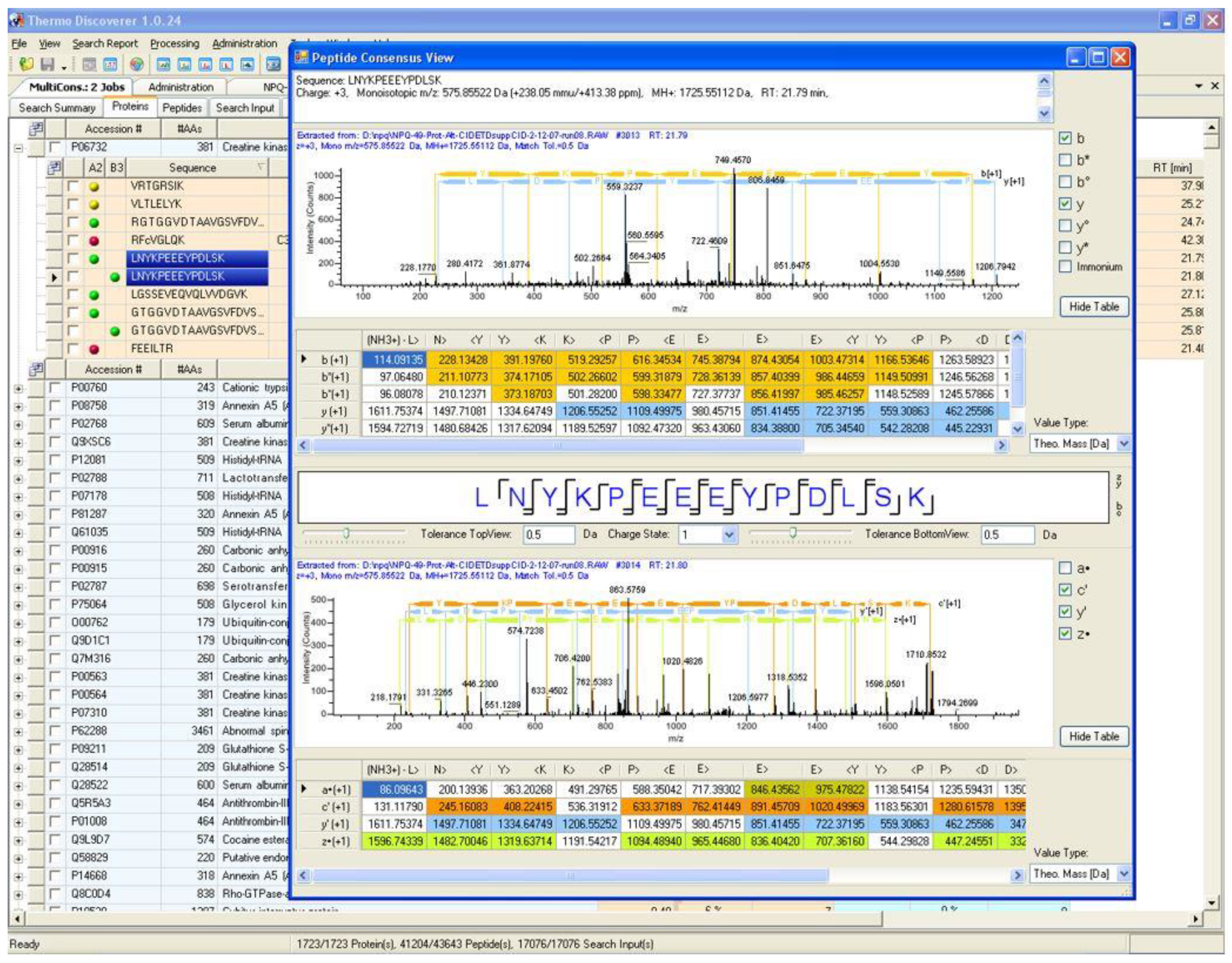
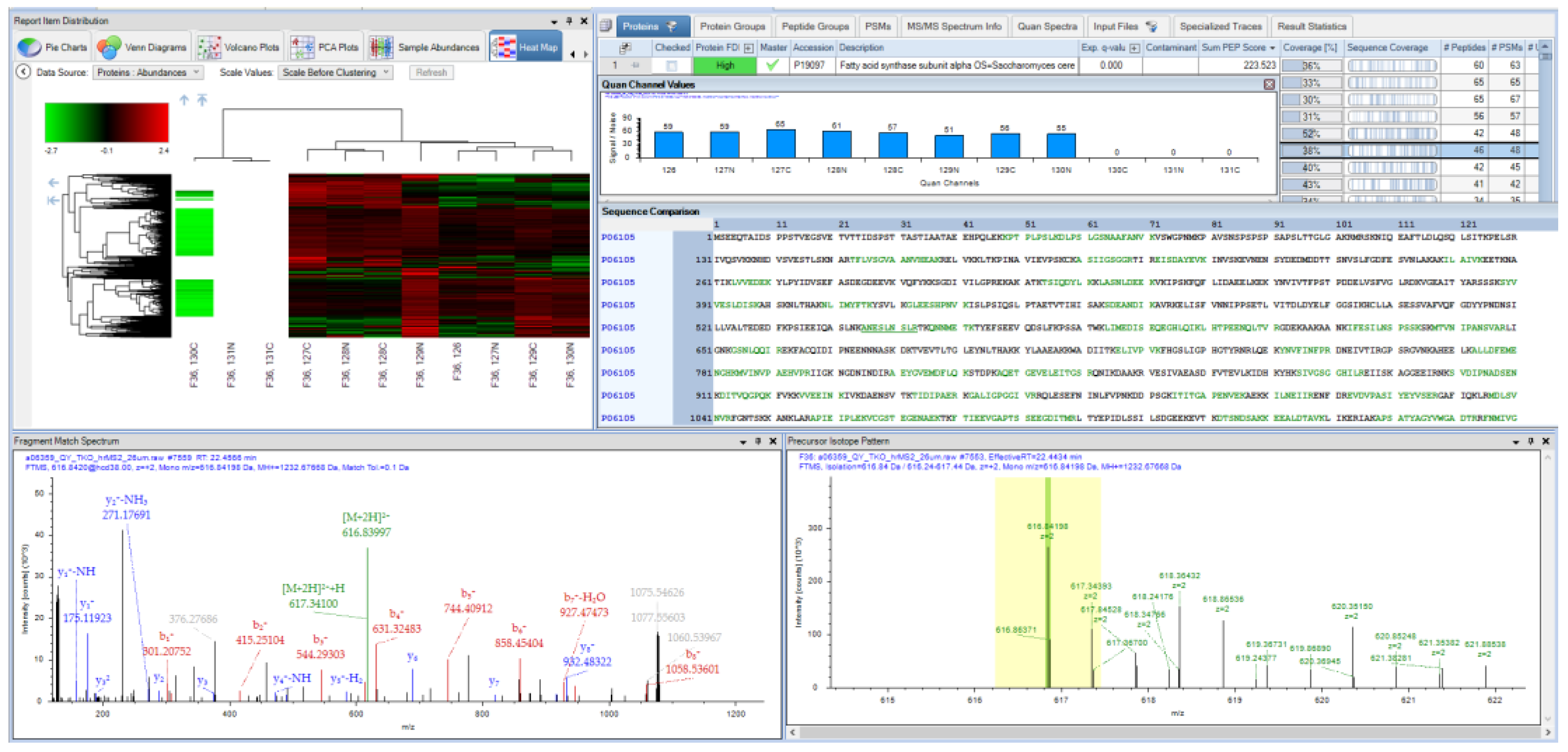
4. A Brief History of Proteome Discoverer Versions and Key Highlights
5. Conclusions: A Biphasic Future for Proteome Discoverer?
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tsiamis, V.; Ienasescu, H.I.; Gabrielaitis, D.; Palmblad, M.; Schwämmle, V.; Ison, J. One Thousand and One Software for Proteomics: Tales of the Toolmakers of Science. J. Proteome Res. 2019, 18, 3580–3585. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Koenig, T.; Menze, B.H.; Kirchner, M.; Monigatti, F.; Parker, K.C.; Patterson, T.; Steen, J.J.; Hamprecht, F.A.; Steen, H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 2008, 7, 3708–3717. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Christoforou, A. Using R and bioconductor for proteomics data analysis. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 42–51. [Google Scholar] [CrossRef]
- Gatto, L.; Breckels, L.M.; Naake, T.; Gibb, S. Visualization of proteomics data using R and Bioconductor. Proteomics 2015, 15, 1375–1389. [Google Scholar] [CrossRef]
- Weisser, H.; Nahnsen, S.; Grossmann, J.; Nilse, L.; Quandt, A.; Brauer, H.; Sturm, M.; Kenar, E.; Kohlbacher, O.; Aebersold, R.; et al. An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 2013, 12, 1628–1644. [Google Scholar] [CrossRef]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets: To the editor. Nat. Biotechnol. 2015, 33, 24. [Google Scholar] [CrossRef]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Kou, Q.; Wu, S.; Liu, X. Systematic Evaluation of Protein Sequence Filtering Algorithms for Proteoform Identification Using Top-Down Mass Spectrometry. Proteomics 2018, 18, 1700306. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.H.; Yang, E.G.; Lee, C.; Lee, J.E. Low-molecular-weight plasma proteome analysis using top-down mass spectrometry. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Kirchner, M.; Steen, J.A.J.; Hamprecht, F.A.; Steen, H. MGFp: An open mascot generic format parser library implementation. J. Proteome Res. 2010, 9, 2762–2763. [Google Scholar] [CrossRef]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Römpp, A.; Neumann, S.; Pizarro, A.D.; et al. mzML—A community standard for mass spectrometry data. Mol. Cell. Proteom. 2011, 10, R110-000133. [Google Scholar] [CrossRef]
- Lin, S.M.; Zhu, L.; Winter, A.Q.; Sasinowski, M.; Kibbe, W.A. What is mzXML good for? Expert Rev. Proteom. 2005, 2, 839–845. [Google Scholar] [CrossRef]
- Turewicz, M.; Deutsch, E.W. Spectra, chromatograms, Metadata: mzML-the standard data format for mass spectrometer output. Methods Mol. Biol. 2011, 696, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Mischerikow, N.; Bandeira, N.; Navarro, J.D.; Wich, L.; Mohammed, S.; Heck, A.J.R.; Pevzner, P.A. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: Applications to database search. Mol. Cell. Proteom. 2010, 9, 2840–2852. [Google Scholar] [CrossRef]
- Rinas, A.; Mali, V.S.; Espino, J.A.; Jones, L.M. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal. Chem. 2016, 88, 10052–10058. [Google Scholar] [CrossRef]
- Chea, E.E.; Rinas, A.; Espino, J.A.; Jones, L.M. Characterizing cellular proteins with in-cell fast photochemical oxidation of proteins. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Carlson, A.; Sinitcyn, P.; Mann, M.; Cox, J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Vaudel, M.; Zhang, B.; Ren, Y.; Wen, B. PDV: An integrative proteomics data viewer. Bioinformatics 2019, 35, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Salinger, A.J.; Dubuke, M.L.; Carmona-Rivera, C.; Maurais, A.J.; Shaffer, S.A.; Weerapana, E.; Thompson, P.R.; Kaplan, M.J. Technical comment on “Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis”. Sci. Immunol. 2020, 5, eaax5672. [Google Scholar] [CrossRef] [PubMed]
- Steckel, A.; Schlosser, G. Citrulline Effect Is a Characteristic Feature of Deiminated Peptides in Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Wang, D.; Wilhelm, M.; Zolg, D.P.; Schmidt, T.; Schnatbaum, K.; Reimer, U.; Pontén, F.; Uhlén, M.; Hahne, H.; et al. Mining the human tissue proteome for protein citrullination. Mol. Cell. Proteom. 2018, 17, 1378–1391. [Google Scholar] [CrossRef]
- König, S. Spectral quality overrides software score—A brief tutorial on the analysis of peptide fragmentation data for mass spectrometry laymen. J. Mass Spectrom. 2020, 56. [Google Scholar] [CrossRef]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Ahmad, S.; Majumder, S.; Jenkins, C.; Orsburn, B. Bolt: A New Age Peptide Search Engine for Comprehensive MS/MS Sequencing Through Vast Protein Databases in Minutes. J. Am. Soc. Mass Spectrom. 2019, 30, 2408–2418. [Google Scholar] [CrossRef]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-independent and Label-free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline. Mol. Cell. Proteomics 2012, 11, 202–214. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Aiyetan, P.; Zhang, B.; Chen, L.; Zhang, Z.; Zhang, H. M2Lite: An Open-source, Light-weight, Pluggable and Fast Proteome Discoverer MSF to mzIdentML Tool. J. Bioinforma. 2014, 1, 40–49. [Google Scholar]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef]
- Ong, S.E.; Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 2007, 1, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Huang, S.Y.; Chow, N.H.; Chen, S.H. Stable-Isotope Dimethyl Labeling for Quantitative Proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J.R. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Spivak, M.; Weston, J.; Bottou, L.; Käll, L.; Noble, W.S. Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J. Proteome Res. 2009, 8, 3737–3745. [Google Scholar] [CrossRef]
- Fondrie, W.E.; Noble, W.S. Machine Learning Strategy That Leverages Large Data sets to Boost Statistical Power in Small-Scale Experiments. J. Proteome Res. 2020, 19, 1267–1274. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villén, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13, 3679–3684. [Google Scholar] [CrossRef] [PubMed]
- Köcher, T.; Pichler, P.; Schutzbier, M.; Stingl, C.; Kaul, A.; Teucher, N.; Hasenfuss, G.; Penninger, J.M.; Mechtler, K. High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap: A new mass spectrometric method combining the benefits of all. J. Proteome Res. 2009, 8, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Pichler, P.; Köcher, T.; Holzmann, J.; Mazanek, M.; Taus, T.; Ammerer, G.; Mechtler, K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ orbitrap. Anal. Chem. 2010, 82, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Hecht, E.S.; Scigelova, M.; Eliuk, S.; Makarov, A. Fundamentals and Advances of Orbitrap Mass Spectrometry. In Encyclopedia of Analytical Chemistry; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Kanawati, B.; Schmitt-Kopplin, P. Fundamentals and Applications of Fourier Transform Mass Spectrometry; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128140147. [Google Scholar]
- Hollingshead, M.G.; Stockwin, L.H.; Alcoser, S.Y.; Newton, D.L.; Orsburn, B.C.; Bonomi, C.A.; Borgel, S.D.; Divelbiss, R.; Dougherty, K.M.; Hager, E.J.; et al. Gene expression profiling of 49 human tumor xenografts from in vitro culture through multiple in vivo passages—Strategies for data mining in support of therapeutic studies. BMC Genom. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Lam, H.; Deutsch, E.W.; Eddes, J.S.; Eng, J.K.; King, N.; Stein, S.E.; Aebersold, R. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7, 655–667. [Google Scholar] [CrossRef]
- Lam, H.; Deutsch, E.W.; Eddes, J.S.; Eng, J.K.; Stein, S.E.; Aebersold, R. Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods 2008, 5, 873–875. [Google Scholar] [CrossRef]
- Zhang, Z.; Burke, M.; Mirokhin, Y.A.; Tchekhovskoi, D.V.; Markey, S.P.; Yu, W.; Chaerkady, R.; Hess, S.; Stein, S.E. Reverse and Random Decoy Methods for False Discovery Rate Estimation in High Mass Accuracy Peptide Spectral Library Searches. J. Proteome Res. 2018, 17, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinforma. 2012, 40, 13.20.1–13.20.14. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Vázquez, J.; Valencia, A.; Tress, M. Analyzing the first drafts of the human proteome. J. Proteome Res. 2014, 13, 3854–3855. [Google Scholar] [CrossRef]
- Veit, J.; Sachsenberg, T.; Chernev, A.; Aicheler, F.; Urlaub, H.; Kohlbacher, O. LFQProfiler and RNPxl: Open-Source Tools for Label-Free Quantification and Protein-RNA Cross-Linking Integrated into Proteome Discoverer. J. Proteome Res. 2016, 15, 3441–3448. [Google Scholar] [CrossRef]
- Fellers, R.T.; Greer, J.B.; Early, B.P.; Yu, X.; Leduc, R.D.; Kelleher, N.L.; Thomas, P.M. ProSight Lite: Graphical software to analyze top-down mass spectrometry data. Proteomics 2015. [Google Scholar] [CrossRef]
- Serang, O.; Käll, L. Solution to Statistical Challenges in Proteomics Is More Statistics, Not Less. J. Proteome Res. 2015, 14, 4099–4103. [Google Scholar] [CrossRef] [PubMed]
- Doblmann, J.; Dusberger, F.; Imre, R.; Hudecz, O.; Stanek, F.; Mechtler, K.; Dürnberger, G. apQuant: Accurate Label-Free Quantification by Quality Filtering. J. Proteome Res. 2018, 18. [Google Scholar] [CrossRef]
- Stadlmann, J.; Hoi, D.M.; Taubenschmid, J.; Mechtler, K.; Penninger, J.M. Analysis of PNGase F-Resistant N-Glycopeptides Using SugarQb for Proteome Discoverer 2.1 Reveals Cryptic Substrate Specificities. Proteomics 2018. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. The cannabis proteome draft map project. Int. J. Mol. Sci. 2020, 21, 965. [Google Scholar] [CrossRef]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Orsburn, B.; Wang, J.; Qu, J. An IonStar Experimental Strategy for MS1 Ion Current-Based Quantification Using Ultrahigh-Field Orbitrap: Reproducible, In-Depth, and Accurate Protein Measurement in Large Cohorts. J. Proteome Res. 2017, 16, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Rijkers, D.T.S.; Post, H.; Heck, A.J.R. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 2015, 12, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lössl, P.; Scheltema, R.; Viner, R.; Heck, A.J.R. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 2017, 8, 15473. [Google Scholar] [CrossRef]
- Klykov, O.; Steigenberger, B.; Pektaş, S.; Fasci, D.; Heck, A.J.R.; Scheltema, R.A. Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 2018, 13, 2964–2990. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Vitek, O.; Nesvizhskii, A.I. A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinform. 2012, 13, S1. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Rinas, A.; Orsburn, B. Direct Measurement of Synchronous Precursor Selection (SPS) Accuracy in Public Proteomics Datasets. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; DIgles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Haw, R.; Hermjakob, H.; D’Eustachio, P.; Stein, L. Reactome pathway analysis to enrich biological discovery in proteomics data sets. Proteomics 2011, 11, 3598–3613. [Google Scholar] [CrossRef] [PubMed]
- Galitzine, C.; Egertson, J.D.; Abbatiello, S.; Henderson, C.M.; Pino, L.K.; MacCoss, M.; Hoofnagle, A.N.; Vitek, O. Nonlinear regression improves accuracy of characterization of multiplexed mass spectrometric assays. Mol. Cell. Proteom. 2018, 17, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef] [PubMed]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsburn, B.C. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. https://doi.org/10.3390/proteomes9010015
Orsburn BC. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes. 2021; 9(1):15. https://doi.org/10.3390/proteomes9010015
Chicago/Turabian StyleOrsburn, Benjamin C. 2021. "Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics" Proteomes 9, no. 1: 15. https://doi.org/10.3390/proteomes9010015
APA StyleOrsburn, B. C. (2021). Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes, 9(1), 15. https://doi.org/10.3390/proteomes9010015