
C1q Complement/Tumor Necrosis Factor-Associated Proteins in Cardiovascular Disease and COVID-19

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
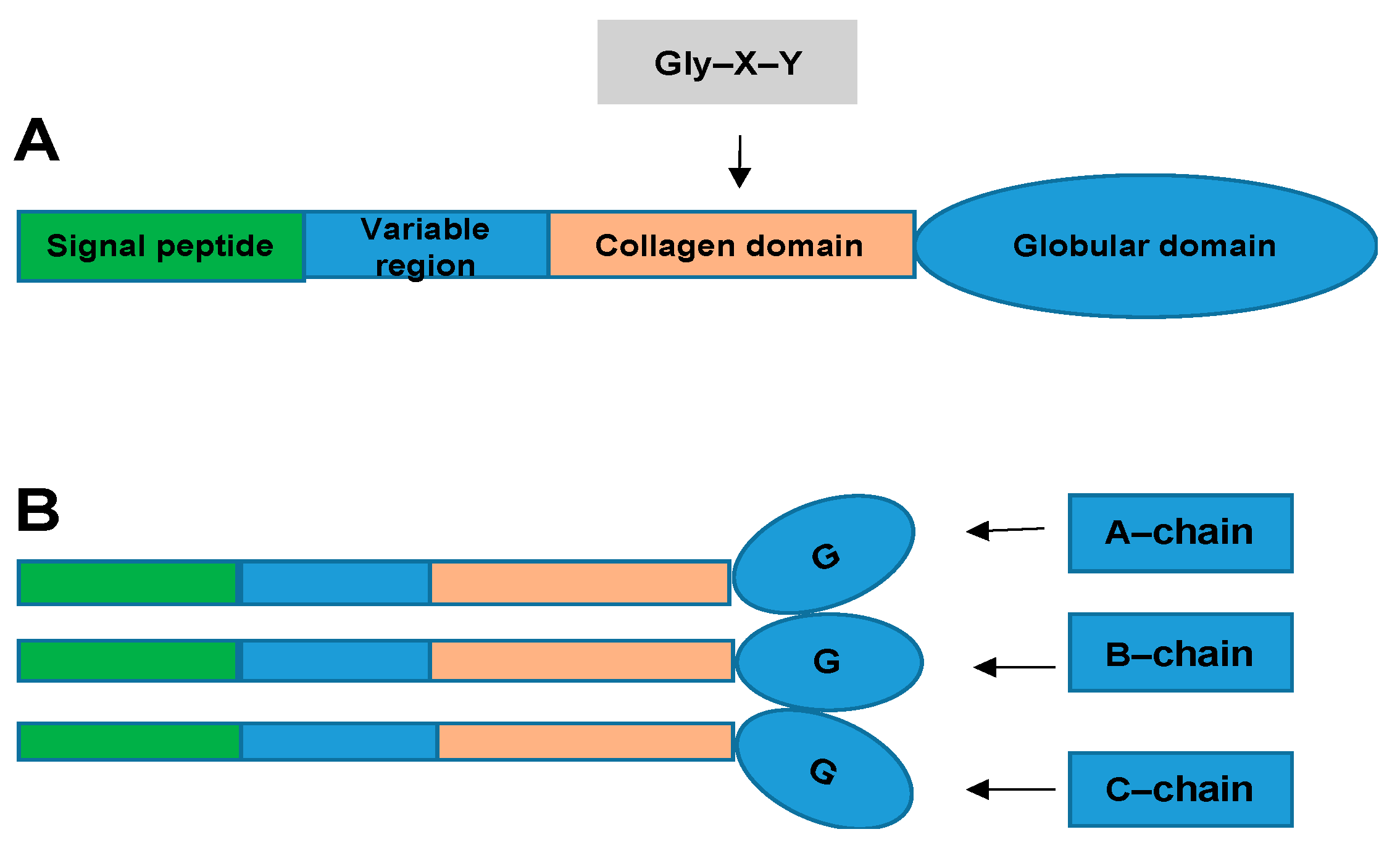
2. What Are CTRPs and How Are They Related to CVD?
3. CTRPs and Vascular Diseases
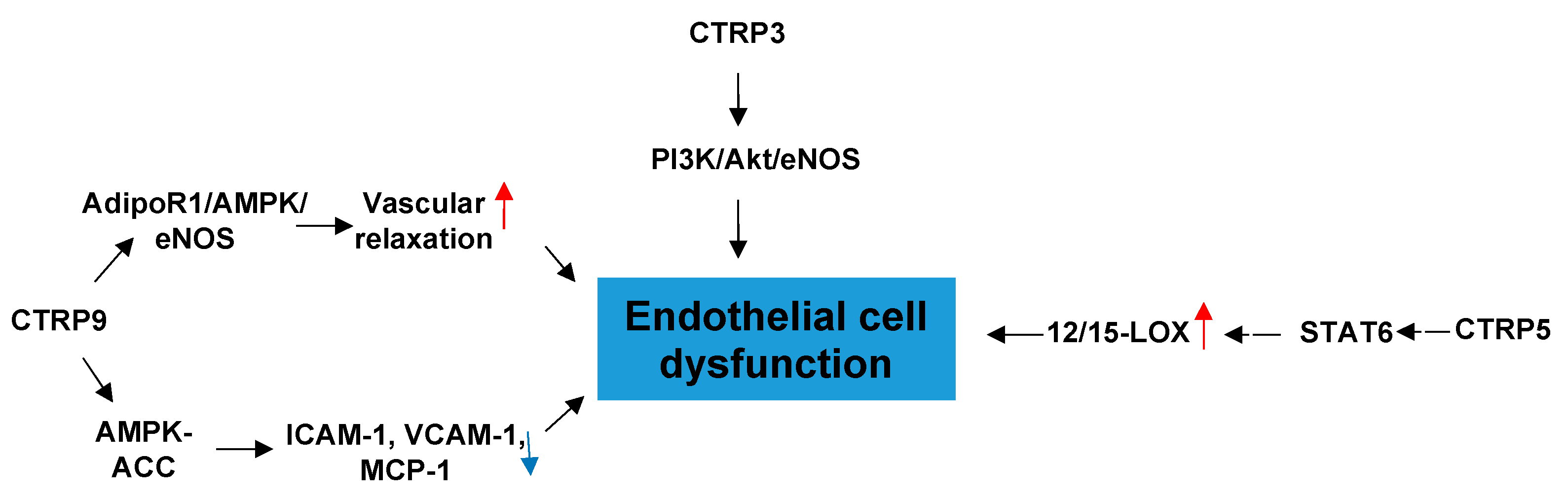
3.1. CTRPs and Endothelial Cell Dysfunction
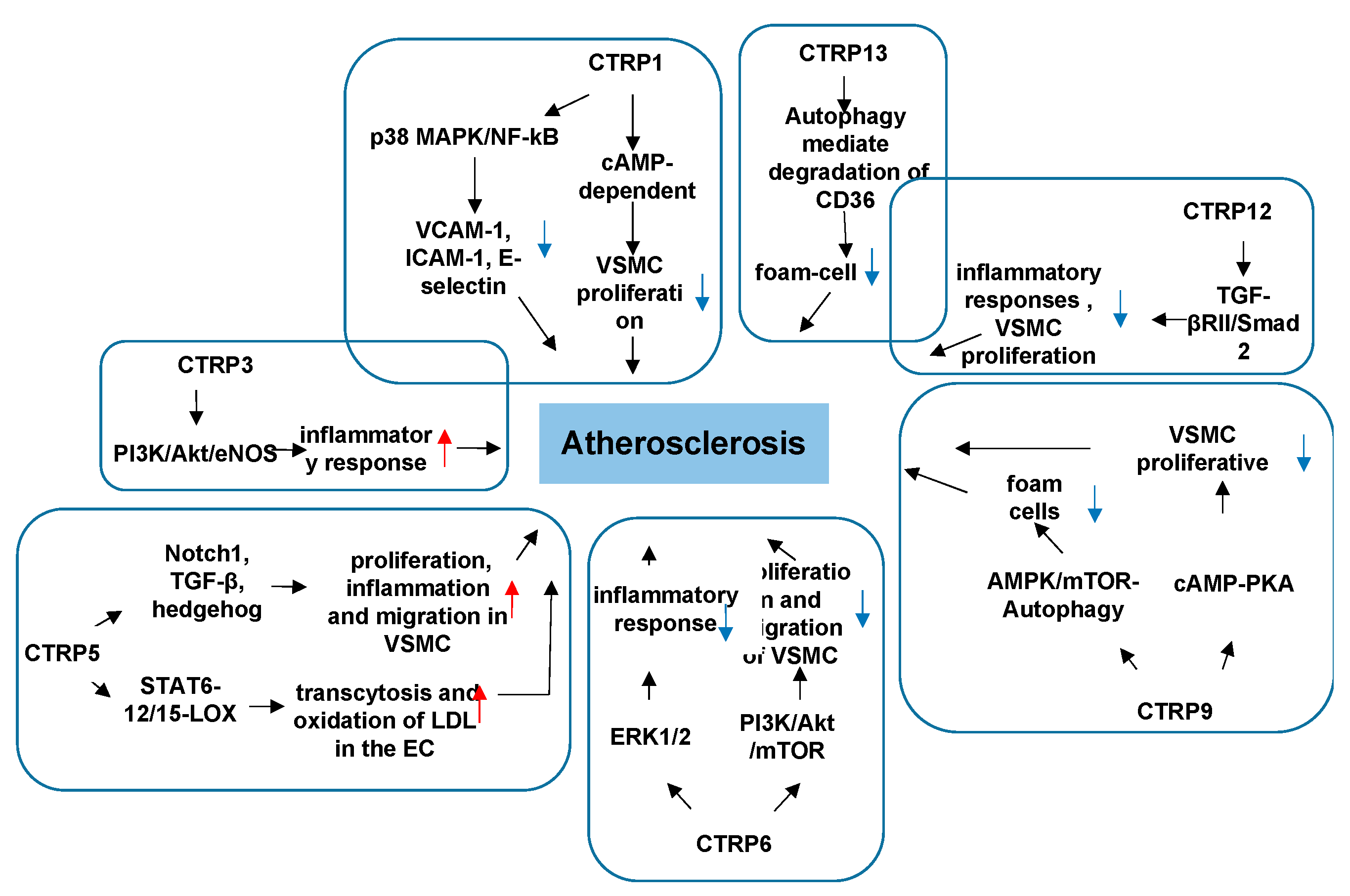
3.2. CTRPs and Atherosclerosis
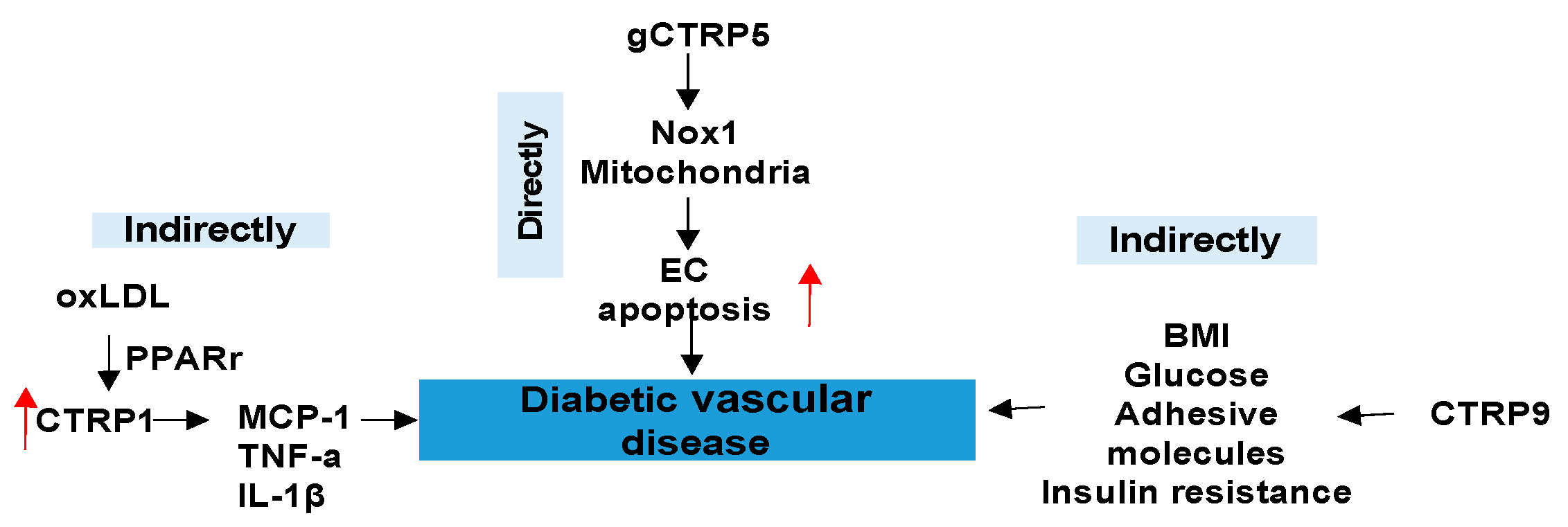
3.3. CTRPs and Diabetic Vascular Disease
4. CTRPs and Cardiac Diseases
4.1. CTRPs and Heart Failure
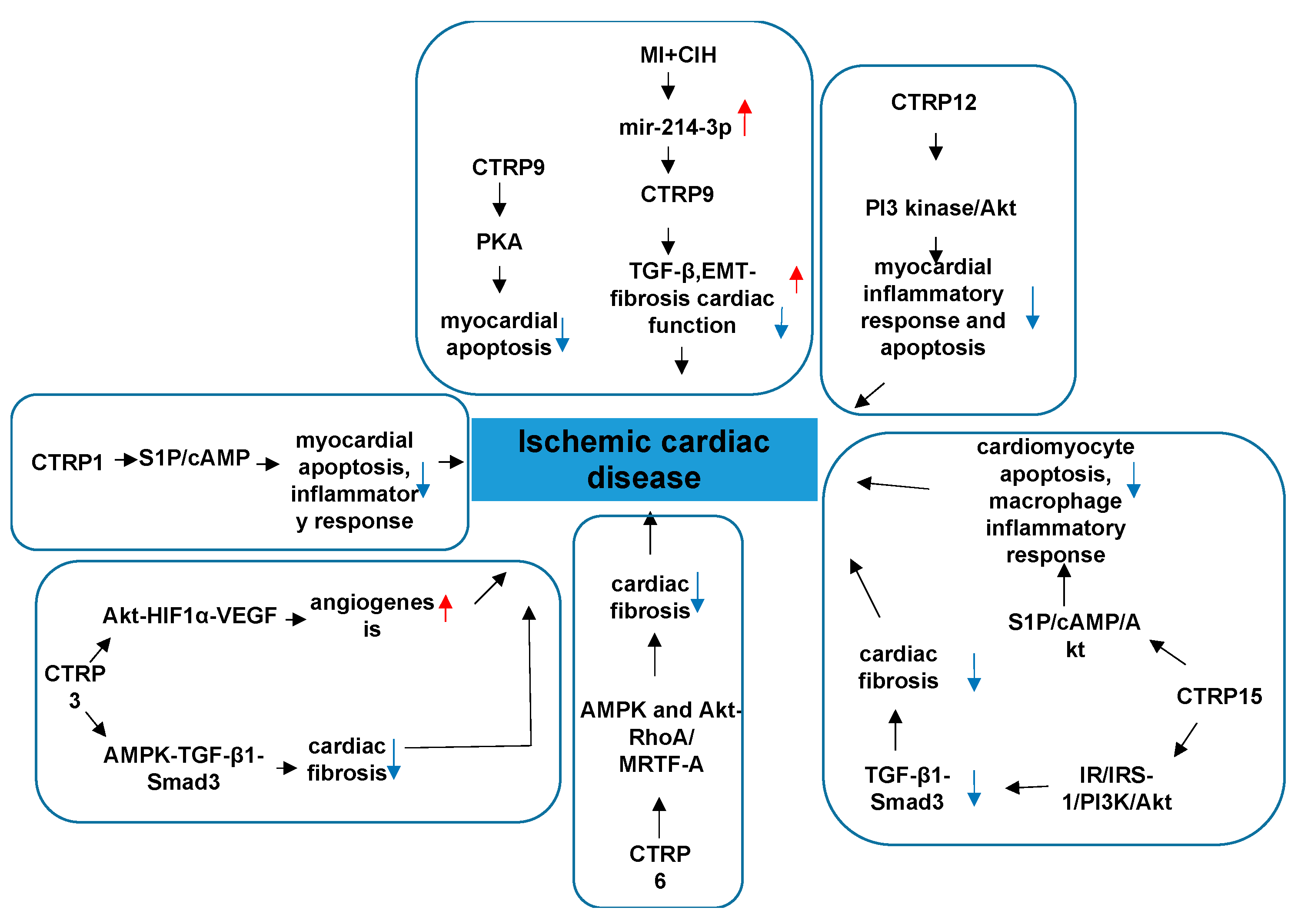
4.2. CTRPs and Ischemic Cardiac Disease
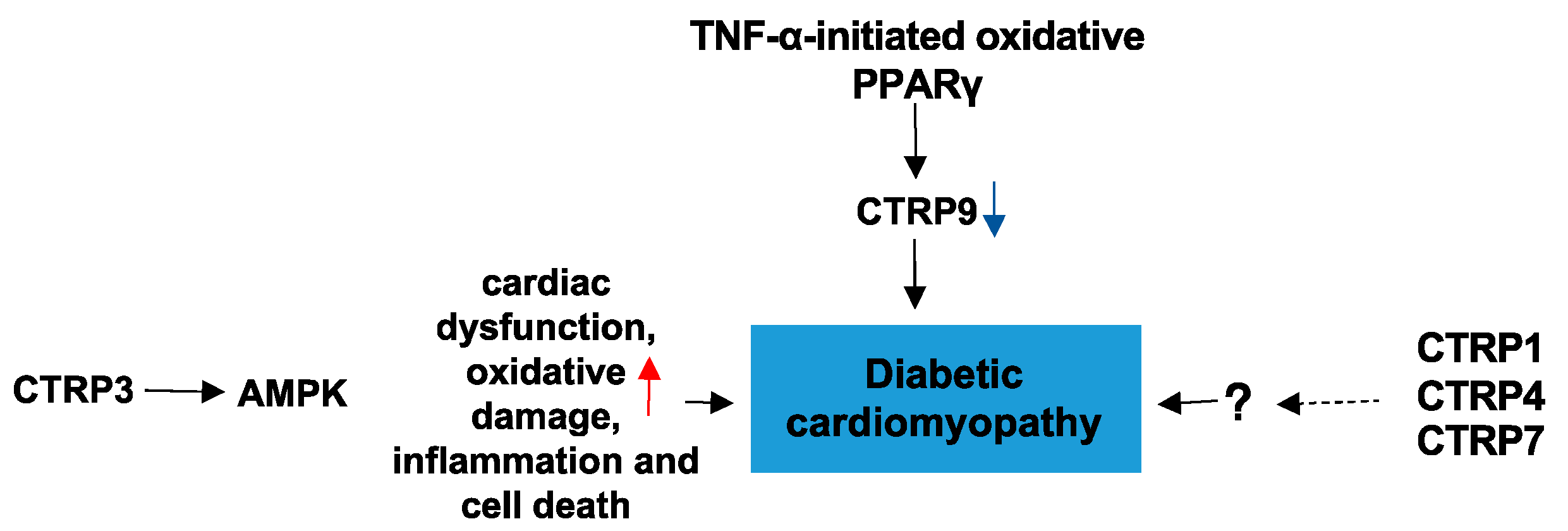
4.3. CTRPs and Diabetic Cardiomyopathy
5. CTRPs and the Role in CVD Patients with COVID-19
5.1. Role of COVID-19 in Cardiovascular Injury
5.2. Diabetes in CVD Patients with COVID-19
5.3. CTRPs in Patients with COVID-19
6. Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Koliaki, C.; Liatis, S.; Kokkinos, A. Obesity and cardiovascular disease: Revisiting an old relationship. Metab. Clin. Exp. 2019, 92, 98–107. [Google Scholar] [CrossRef]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stachel, A. Obesity in Patients Younger Than 60 Years Is a Risk Factor for COVID-19 Hospital Admission. Clin. Infect. Dis. 2020, 71, 896–897. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Wong, G.W.; Wang, J.; Hug, C.; Tsao, T.S.; Lodish, H.F. A family of Acrp30/adiponectin structural and functional paralogs. Proc. Natl. Acad. Sci. USA 2004, 101, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561. [Google Scholar] [CrossRef]
- Ghai, R.; Waters, P.; Roumenina, L.T.; Gadjeva, M.; Kojouharova, M.S.; Reid, K.B.; Sim, R.B.; Kishore, U. C1q and its growing family. Immunobiology 2007, 212, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Schaffler, A.; Buechler, C. CTRP family: Linking immunity to metabolism. Trends Endocrinol. Metab. TEM 2012, 23, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Shanaki, M.; Shabani, P.; Goudarzi, A.; Omidifar, A.; Bashash, D.; Emamgholipour, S. The C1q/TNF-related proteins (CTRPs) in pathogenesis of obesity-related metabolic disorders: Focus on type 2 diabetes and cardiovascular diseases. Life Sci. 2020, 256, 117913. [Google Scholar] [CrossRef]
- Si, Y.; Fan, W.; Sun, L. A Review of the Relationship Between CTRP Family and Coronary Artery Disease. Curr. Atheroscler. Rep. 2020, 22, 22. [Google Scholar] [CrossRef]
- Chen, L.; Qin, L.; Liu, X.; Meng, X. CTRP3 Alleviates Ox-LDL-Induced Inflammatory Response and Endothelial Dysfunction in Mouse Aortic Endothelial Cells by Activating the PI3K/Akt/eNOS Pathway. Inflammation 2019, 42, 1350–1359. [Google Scholar] [CrossRef]
- Li, C.; Chen, J.W.; Liu, Z.H.; Shen, Y.; Ding, F.H.; Gu, G.; Liu, J.; Qiu, J.P.; Gao, J.; Zhang, R.Y.; et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis 2018, 278, 197–209. [Google Scholar] [CrossRef]
- Su, H.; Yuan, Y.; Wang, X.M.; Lau, W.B.; Wang, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.L. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res. Cardiol. 2013, 108, 315. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Krawczyk, S.A.; Kitidis-Mitrokostas, C.; Ge, G.; Spooner, E.; Hug, C.; Gimeno, R.; Lodish, H.F. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009, 23, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Lau, W.B.; Su, H.; Sun, Y.; Yi, W.; Du, Y.; Christopher, T.; Lopez, B.; Wang, Y.; Ma, X.L. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E891–E898. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Tan, S.Y.; Wong, G.W. Metabolic function of the CTRP family of hormones. Rev. Endocr. Metab. Disorders 2014, 15, 111–123. [Google Scholar] [CrossRef]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Lee, Y.L.; Seol, S.M.; Yoon, H.K.; Kang, S.W.; Lee, W.J.; Park, J.Y. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol. Cell. Endocrinol. 2016, 419, 235–243. [Google Scholar] [CrossRef]
- Cheng, L.; Li, B.; Chen, X.; Su, J.; Wang, H.; Yu, S.; Zheng, Q. CTRP9 induces mitochondrial biogenesis and protects high glucose-induced endothelial oxidative damage via AdipoR1 -SIRT1- PGC-1α activation. Biochem. Biophys. Res. Commun. 2016, 477, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Yuan, Y.; Yi, W.; Lau, W.B.; Wang, Y.; Wang, X.; Sun, Y.; Lopez, B.L.; Christopher, T.A.; Peterson, J.M.; et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2616–2623. [Google Scholar] [CrossRef]
- Liu, J.; Meng, Z.; Gan, L.; Guo, R.; Gao, J.; Liu, C.; Zhu, D.; Liu, D.; Zhang, L.; Zhang, Z.; et al. C1q/TNF-related protein 5 contributes to diabetic vascular endothelium dysfunction through promoting Nox-1 signaling. Redox Biol. 2020, 34, 101476. [Google Scholar] [CrossRef]
- Wang, X.Q.; Liu, Z.H.; Xue, L.; Lu, L.; Gao, J.; Shen, Y.; Yang, K.; Chen, Q.J.; Zhang, R.Y.; Shen, W.F. C1q/TNF-related protein 1 links macrophage lipid metabolism to inflammation and atherosclerosis. Atherosclerosis 2016, 250, 38–45. [Google Scholar] [CrossRef]
- Uemura, Y.; Shibata, R.; Ohashi, K.; Enomoto, T.; Kambara, T.; Yamamoto, T.; Ogura, Y.; Yuasa, D.; Joki, Y.; Matsuo, K.; et al. Adipose-derived factor CTRP9 attenuates vascular smooth muscle cell proliferation and neointimal formation. FASEB J. 2013, 27, 25–33. [Google Scholar] [CrossRef]
- Majidi, Z.; Emamgholipour, S.; Omidifar, A.; Rahmani Fard, S.; Poustchi, H.; Shanaki, M. The circulating levels of CTRP1 and CTRP5 are associated with obesity indices and carotid intima-media thickness (cIMT) value in patients with type 2 diabetes: A preliminary study. Diabetol. Metab. Syndr. 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Moradi, N.; Fadaei, R.; Rashidbeygi, E.; Bagheri Kargasheh, F.; Malek, M.; Shokoohi Nahrkhalaji, A.; Fallah, S. Evaluation of changing the pattern of CTRP5 and inflammatory markers levels in patients with coronary artery disease and type 2 diabetes mellitus. Arch. Physiol. Biochem. 2020, 1–6. [Google Scholar] [CrossRef]
- Kanemura, N.; Shibata, R.; Ohashi, K.; Ogawa, H.; Hiramatsu-Ito, M.; Enomoto, T.; Yuasa, D.; Ito, M.; Hayakawa, S.; Otaka, N.; et al. C1q/TNF-related protein 1 prevents neointimal formation after arterial injury. Atherosclerosis 2017, 257, 138–145. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, R.Y.; Wang, X.Q.; Liu, Z.H.; Shen, Y.; Ding, F.H.; Meng, H.; Wang, L.J.; Yan, X.X.; Yang, K.; et al. C1q/TNF-related protein-1: An adipokine marking and promoting atherosclerosis. Eur. Heart J. 2016, 37, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Wei, Z.; Wong, G.W. C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J. Biol. Chem. 2010, 285, 39691–39701. [Google Scholar] [CrossRef]
- Kopp, A.; Bala, M.; Buechler, C.; Falk, W.; Gross, P.; Neumeier, M.; Scholmerich, J.; Schaffler, A. C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology 2010, 151, 5267–5278. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, M.J.; Park, E.J.; Choi, Y.J.; Park, S.Y. C1qTNF-related protein-6 mediates fatty acid oxidation via the activation of the AMP-activated protein kinase. FEBS Lett. 2010, 584, 968–972. [Google Scholar] [CrossRef]
- Wei, Z.; Peterson, J.M.; Lei, X.; Cebotaru, L.; Wolfgang, M.J.; Baldeviano, G.C.; Wong, G.W. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J. Biol. Chem. 2012, 287, 10301–10315. [Google Scholar] [CrossRef]
- Fadaei, R.; Moradi, N.; Kazemi, T.; Chamani, E.; Azdaki, N.; Moezibady, S.A.; Shahmohamadnejad, S.; Fallah, S. Decreased serum levels of CTRP12/adipolin in patients with coronary artery disease in relation to inflammatory cytokines and insulin resistance. Cytokine 2019, 113, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Ohashi, K.; Ito, M.; Shibata, R.; Kanemura, N.; Yuasa, D.; Kambara, T.; Matsuo, K.; Hayakawa, S.; Hiramatsu-Ito, M.; et al. Adipolin/CTRP12 protects against pathological vascular remodelling through suppression of smooth muscle cell growth and macrophage inflammatory response. Cardiovasc. Res. 2020, 116, 237–249. [Google Scholar] [CrossRef]
- Lutgens, E.; Gijbels, M.; Smook, M.; Heeringa, P.; Gotwals, P.; Koteliansky, V.E.; Daemen, M.J. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, C.; Zhang, R.Y.; Zhang, Q.; Shen, W.F.; Ding, F.H.; Lu, L. Association of increased serum CTRP5 levels with in-stent restenosis after coronary drug-eluting stent implantation: CTRP5 promoting inflammation, migration and proliferation in vascular smooth muscle cells. Int. J. Cardiol. 2017, 228, 129–136. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, W.; Park, E.J.; Park, S.Y. C1qTNF-related protein-6 increases the expression of interleukin-10 in macrophages. Mol. Cells 2010, 30, 59–64. [Google Scholar] [CrossRef]
- Dong, X.; Hu, H.; Fang, Z.; Cui, J.; Liu, F. CTRP6 inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and migration. Biomed. Pharmacother. 2018, 103, 844–850. [Google Scholar] [CrossRef]
- Li, J.; Zhang, P.; Li, T.; Liu, Y.; Zhu, Q.; Chen, T.; Liu, T.; Huang, C.; Zhang, J.; Zhang, Y.; et al. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem. Biophys. Res. Commun. 2015, 458, 890–895. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, C.; Li, J.; Li, T.; Guo, H.; Liu, T.; Li, N.; Zhu, Q.; Guo, Y. Globular CTRP9 inhibits oxLDL-induced inflammatory response in RAW 264.7 macrophages via AMPK activation. Mol. Cell. Biochem. 2016, 417, 67–74. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Q.; Zhang, H.; Wang, X.D.; Chen, S.Y.; Yang, Y.; Lv, H.; Hou, J.B.; Yu, B. C1q/TNF-Related Protein 9 Inhibits THP-1 Macrophage Foam Cell Formation by Enhancing Autophagy. J. Cardiovasc. Pharmacol. 2018, 72, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gong, X.; Ni, S.; Wang, Y.; Zhu, L.; Ji, N. C1q/TNF-related protein-9 attenuates atherosclerosis through AMPK-NLRP3 inflammasome singling pathway. Int. Immunopharmacol. 2019, 77, 105934. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, W.; Liang, M.; Huang, D.; Huang, K. CTRP13 inhibits atherosclerosis via autophagy-lysosome-dependent degradation of CD36. FASEB J. Off. Publ. Federation Am. Soc. Exp. Biol. 2019, 33, 2290–2300. [Google Scholar] [CrossRef]
- Fadaei, R.; Moradi, N.; Baratchian, M.; Aghajani, H.; Malek, M.; Fazaeli, A.A.; Fallah, S. Association of C1q/TNF-Related Protein-3 (CTRP3) and CTRP13 Serum Levels with Coronary Artery Disease in Subjects with and without Type 2 Diabetes Mellitus. PLoS ONE 2016, 11, e0168773. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Wei, Z.; Peterson, J.M.; Wong, G.W. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): Activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J. Biol. Chem. 2011, 286, 15652–15665. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Jang, J.E.; Leem, J.; Lee, Y.L.; Seol, S.M.; Yoon, H.K.; Lee, W.J.; Park, J.Y. Association of serum C1q/TNF-related protein-9 concentration with arterial stiffness in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E2477–E2484. [Google Scholar] [CrossRef][Green Version]
- Moradi, N.; Fadaei, R.; Emamgholipour, S.; Kazemian, E.; Panahi, G.; Vahedi, S.; Saed, L.; Fallah, S. Association of circulating CTRP9 with soluble adhesion molecules and inflammatory markers in patients with type 2 diabetes mellitus and coronary artery disease. PLoS ONE 2018, 13, e0192159. [Google Scholar] [CrossRef]
- Schmid, A.; Kopp, A.; Aslanidis, C.; Wabitsch, M.; Muller, M.; Schaffler, A. Regulation and function of C1Q/TNF-related protein-5 (CTRP-5) in the context of adipocyte biology. Exp. Clin. Endocrinol. Diabetes 2013, 121, 310–317. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Ryu, H.S.; Pak, Y.K.; Park, K.S.; Lee, H.K.; Lee, W. C1q tumor necrosis factor alpha-related protein isoform 5 is increased in mitochondrial DNA-depleted myocytes and activates AMP-activated protein kinase. J. Biol. Chem. 2009, 284, 27780–27789. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Kasher Meron, M.; Xu, S.; Glesby, M.J.; Qi, Q.; Hanna, D.B.; Anastos, K.; Kaplan, R.C.; Kizer, J.R. C1q/TNF-Related Proteins, HIV and HIV-Associated Factors, and Cardiometabolic Phenotypes in Middle-Aged Women. AIDS Res. Hum. Retroviruses 2019, 35, 1054–1064. [Google Scholar] [CrossRef]
- Ma, Z.G.; Yuan, Y.P.; Zhang, X.; Xu, S.C.; Kong, C.Y.; Song, P.; Li, N.; Tang, Q.Z. C1q-tumour necrosis factor-related protein-3 exacerbates cardiac hypertrophy in mice. Cardiovasc. Res. 2019, 115, 1067–1077. [Google Scholar] [CrossRef]
- Appari, M.; Breitbart, A.; Brandes, F.; Szaroszyk, M.; Froese, N.; Korf-Klingebiel, M.; Mohammadi, M.M.; Grund, A.; Scharf, G.M.; Wang, H.; et al. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ. Res. 2017, 120, 66–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Liu, J.; Guo, R.; Yan, Z.; Liu, W.; Lau, W.B.; Jiao, X.; Cao, J.; Xu, K.; et al. Implications of C1q/TNF-related protein superfamily in patients with coronary artery disease. Sci. Rep. 2020, 10, 878. [Google Scholar] [CrossRef]
- Yuasa, D.; Ohashi, K.; Shibata, R.; Mizutani, N.; Kataoka, Y.; Kambara, T.; Uemura, Y.; Matsuo, K.; Kanemura, N.; Hayakawa, S.; et al. C1q/TNF-related protein-1 functions to protect against acute ischemic injury in the heart. FASEB J. 2016, 30, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Sun, Y.; Yuan, Y.; Lau, W.B.; Zheng, Q.; Wang, X.; Wang, Y.; Shang, X.; Gao, E.; Koch, W.J.; et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation 2012, 125, 3159–3169. [Google Scholar] [CrossRef]
- Wu, D.; Lei, H.; Wang, J.Y.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Li, L.; Wu, L.L. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J. Mol. Med. 2015, 93, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Wu, D.; Wang, J.Y.; Li, L.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Wu, L.L. C1q/tumor necrosis factor-related protein-6 attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF-A pathway and inhibiting myofibroblast differentiation. Basic Res. Cardiol. 2015, 110, 35. [Google Scholar] [CrossRef]
- Takikawa, T.; Ohashi, K.; Ogawa, H.; Otaka, N.; Kawanishi, H.; Fang, L.; Ozaki, Y.; Eguchi, S.; Tatsumi, M.; Takefuji, M.; et al. Adipolin/C1q/Tnf-related protein 12 prevents adverse cardiac remodeling after myocardial infarction. PLoS ONE 2020, 15, e0243483. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J. Biol. Chem. 2012, 287, 11968–11980. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Lei, X.; Tan, S.Y.; Stanson, K.P.; Wei, Z.; Wong, G.W. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J. Biol. Chem. 2013, 288, 36073–36082. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef]
- Jiang, X.; Gao, M.; Chen, Y.; Liu, J.; Qi, S.; Ma, J.; Zhang, Z.; Xu, Y. EPO-dependent induction of erythroferrone drives hepcidin suppression and systematic iron absorption under phenylhydrazine-induced hemolytic anemia. Blood Cells Mol. Dis. 2016, 58, 45–51. [Google Scholar] [CrossRef]
- Latour, C.; Wlodarczyk, M.F.; Jung, G.; Gineste, A.; Blanchard, N.; Ganz, T.; Roth, M.P.; Coppin, H.; Kautz, L. Erythroferrone contributes to hepcidin repression in a mouse model of malarial anemia. Haematologica 2017, 102, 60–68. [Google Scholar] [CrossRef]
- Ouchi, N.; Walsh, K. Cardiovascular and metabolic regulation by the adiponectin/C1q/tumor necrosis factor-related protein family of proteins. Circulation 2012, 125, 3066–3068. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, C.L.; Xiang, R.L.; Wu, L.L.; Li, L. CTRP15 derived from cardiac myocytes attenuates TGFβ1-induced fibrotic response in cardiac fibroblasts. Cardiovasc. Drugs Ther. 2020, 34, 591–604. [Google Scholar] [CrossRef]
- Otaka, N.; Shibata, R.; Ohashi, K.; Uemura, Y.; Kambara, T.; Enomoto, T.; Ogawa, H.; Ito, M.; Kawanishi, H.; Maruyama, S.; et al. Myonectin Is an Exercise-Induced Myokine That Protects the Heart From Ischemia-Reperfusion Injury. Circ. Res. 2018, 123, 1326–1338. [Google Scholar] [CrossRef]
- Wang, J.; Hang, T.; Cheng, X.M.; Li, D.M.; Zhang, Q.G.; Wang, L.J.; Peng, Y.P.; Gong, J.B. Associations of C1q/TNF-Related Protein-9 Levels in Serum and Epicardial Adipose Tissue with Coronary Atherosclerosis in Humans. BioMed. Res. Int. 2015, 2015, 971683. [Google Scholar] [CrossRef]
- Sun, Y.; Yi, W.; Yuan, Y.; Lau, W.B.; Yi, D.; Wang, X.; Wang, Y.; Su, H.; Wang, X.; Gao, E.; et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013, 128, S113–S120. [Google Scholar] [CrossRef]
- Du, Y.; Wang, X.; Li, L.; Hao, W.; Zhang, H.; Li, Y.; Qin, Y.; Nie, S.; Christopher, T.A.; Lopez, B.L.; et al. miRNA-Mediated Suppression of a Cardioprotective Cardiokine as a Novel Mechanism Exacerbating Post-MI Remodeling by Sleep Breathing Disorders. Circ. Res. 2020, 126, 212–228. [Google Scholar] [CrossRef]
- Liu, M.; Li, W.; Wang, H.; Yin, L.; Ye, B.; Tang, Y.; Huang, C. CTRP9 Ameliorates Atrial Inflammation, Fibrosis, and Vulnerability to Atrial Fibrillation in Post-Myocardial Infarction Rats. J. Am. Heart Assoc. 2019, 8, e013133. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Ma, Z.G.; Yuan, Y.P.; Xu, S.C.; Wei, W.Y.; Xu, C.R.; Zhang, X.; Wu, Q.Q.; Liao, H.H.; Ni, J.; Tang, Q.Z. CTRP3 attenuates cardiac dysfunction, inflammation, oxidative stress and cell death in diabetic cardiomyopathy in rats. Diabetologia 2017, 60, 1126–1137. [Google Scholar] [CrossRef]
- Sun, P.; Lu, X.; Xu, C.; Sun, W.; Pan, B. Understanding of COVID-19 based on current evidence. J. Med. Virol. 2020, 92, 548–551. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Ann. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Bansal, M. Cardiovascular disease and COVID-19. Diabetes Metab. Syndrome 2020, 14, 247–250. [Google Scholar] [CrossRef]
- Kochi, A.N.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID-19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K. Correction to: Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 1294–1297. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612–1614. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K. Obesity, vitamin D deficiency and old age a serious combination with respect to coronavirus disease-2019 severity and outcome. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 18–24. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Saner, C.; Riksen, N.P.; Netea, M.G.; Sabin, M.A.; Saffery, R.; Stienstra, R.; Burgner, D.P. Trained Immunity: Linking Obesity and Cardiovascular Disease across the Life-Course? Trends Endocrinol. Metab. TEM 2020, 31, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Carlson, F.R., Jr.; Bosukonda, D.; Keck, P.C.; Carlson, W.D. Multiorgan Damage in Patients With COVID-19: Is the TGF-beta/BMP Pathway the Missing Link? JACC. Basic Transl. sci. 2020, 5, 1145–1148. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Lombardi, C.M.; Carubelli, V.; Iorio, A.; Inciardi, R.M.; Bellasi, A.; Canale, C.; Camporotondo, R.; Catagnano, F.; Dalla Vecchia, L.A.; Giovinazzo, S.; et al. Association of Troponin Levels With Mortality in Italian Patients Hospitalized With Coronavirus Disease 2019: Results of a Multicenter Study. JAMA Cardiol. 2020, 5, 1274–1280. [Google Scholar] [CrossRef]
- Sakamoto, A.; Kawakami, R.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Kutys, R.; Guo, L.; Mori, M.; Cornelissen, A.; Sato, Y.; et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Inciardi, R.M.; Lombardi, C.M.; Tedino, C.; Agostoni, P.; Ameri, P.; Barbieri, L.; Bellasi, A.; Camporotondo, R.; Canale, C.; et al. Impact of heart failure on the clinical course and outcomes of patients hospitalized for COVID-19. Results of the Cardio-COVID-Italy multicentre study. Eur. J. Heart Fail 2020, 22, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Messina, G.; Polito, R.; Monda, V.; Cipolloni, L.; Di Nunno, N.; Di Mizio, G.; Murabito, P.; Carotenuto, M.; Messina, A.; Pisanelli, D.; et al. Functional Role of Dietary Intervention to Improve the Outcome of COVID-19: A Hypothesis of Work. Int. J. Mol. Sci. 2020, 21, 3104. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Meng, Z.; Gao, J.; Liu, C.; Wang, J.; Guo, R.; Zhao, J.; Lopez, B.; Christopher, T.; Lee, D.; et al. C1q Complement/Tumor Necrosis Factor-Associated Proteins in Cardiovascular Disease and COVID-19. Proteomes 2021, 9, 12. https://doi.org/10.3390/proteomes9010012
Xie Y, Meng Z, Gao J, Liu C, Wang J, Guo R, Zhao J, Lopez B, Christopher T, Lee D, et al. C1q Complement/Tumor Necrosis Factor-Associated Proteins in Cardiovascular Disease and COVID-19. Proteomes. 2021; 9(1):12. https://doi.org/10.3390/proteomes9010012
Chicago/Turabian StyleXie, Yaoli, Zhijun Meng, Jia Gao, Caihong Liu, Jing Wang, Rui Guo, Jianli Zhao, Bernard Lopez, Theodore Christopher, Daniel Lee, and et al. 2021. "C1q Complement/Tumor Necrosis Factor-Associated Proteins in Cardiovascular Disease and COVID-19" Proteomes 9, no. 1: 12. https://doi.org/10.3390/proteomes9010012
APA StyleXie, Y., Meng, Z., Gao, J., Liu, C., Wang, J., Guo, R., Zhao, J., Lopez, B., Christopher, T., Lee, D., Ma, X., & Wang, Y. (2021). C1q Complement/Tumor Necrosis Factor-Associated Proteins in Cardiovascular Disease and COVID-19. Proteomes, 9(1), 12. https://doi.org/10.3390/proteomes9010012