Differential Protein Expression in Striatal D1- and D2-Dopamine Receptor-Expressing Medium Spiny Neurons

 , , ,
, , ,  and
and
Abstract
1. Introduction
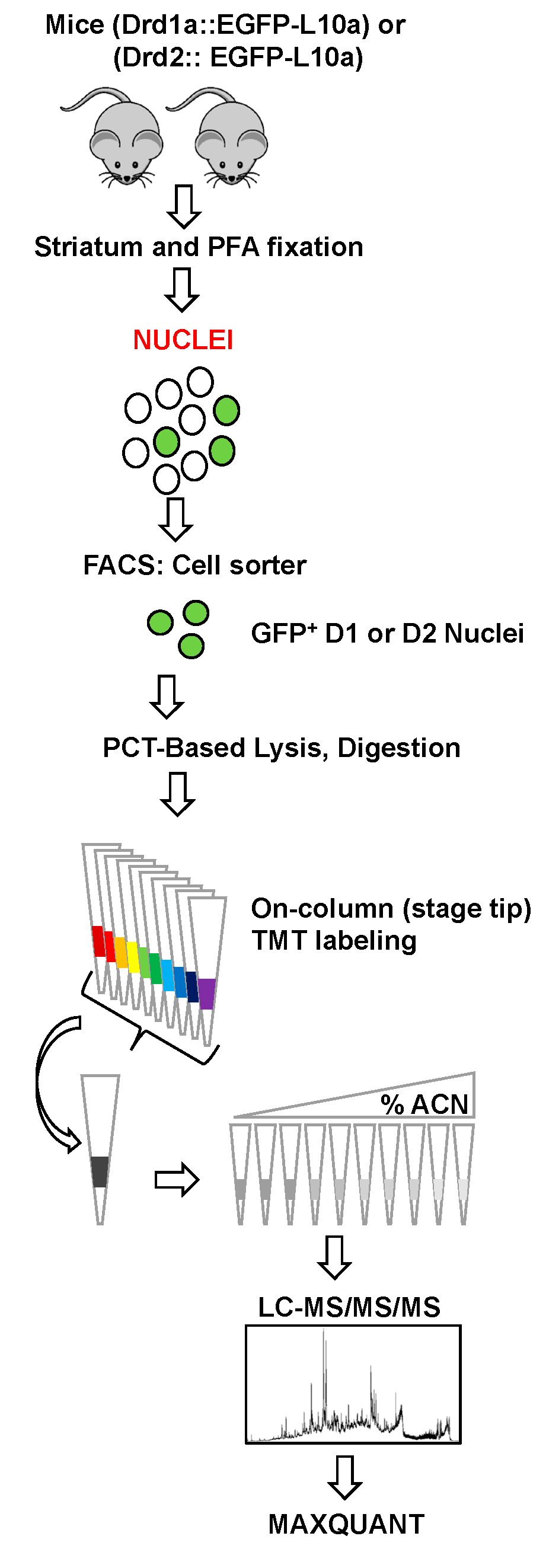
2. Materials and Methods
2.1. Animals
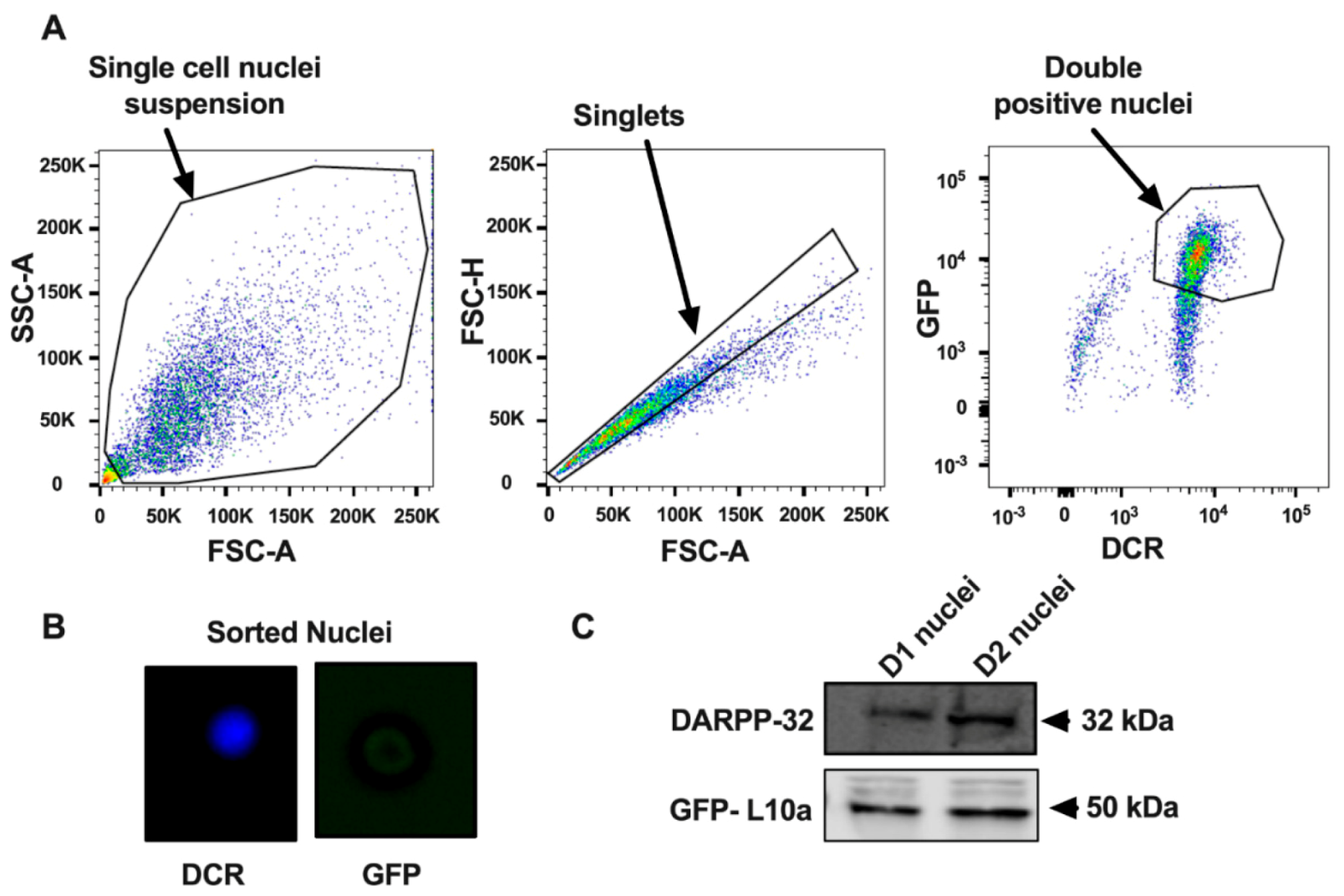
2.2. Nuclei Isolation and FANS
2.3. Immunoblotting of Nuclei
2.4. Sample Preparation for LC-MS/MS/MS
2.4.1. Pressure-Cycling Technology (PCT)-Based Lysis and Digestion
2.4.2. On-Column TMT Labeling
2.4.3. Stage Tip bSDB Fractionation
2.5. Data Acquisition
2.6. Data Processing and Analysis
3. Results
3.1. Purification of Nuclei
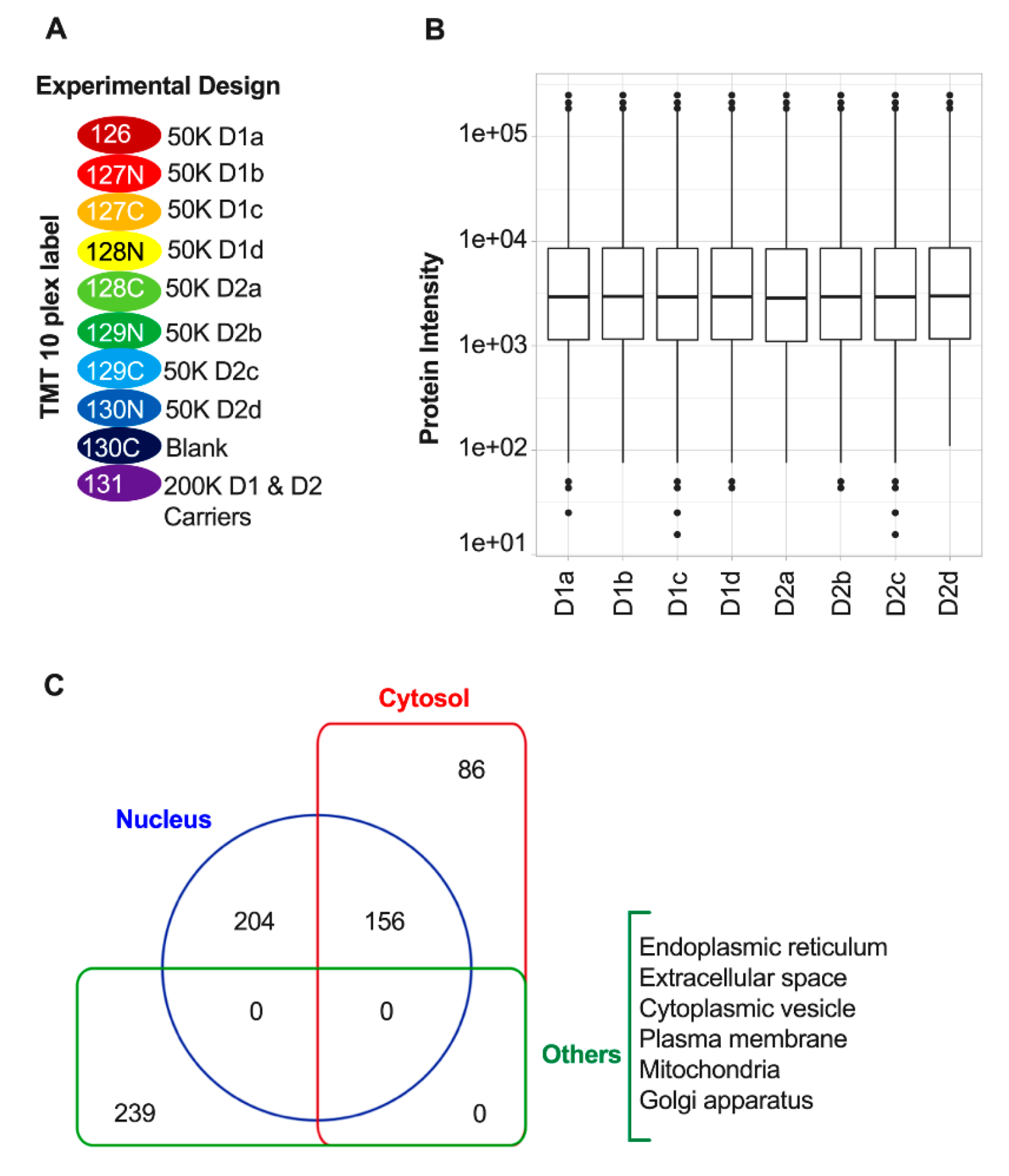
3.2. Quantitative Proteomic Profiling of D1 and D2 Nuclei
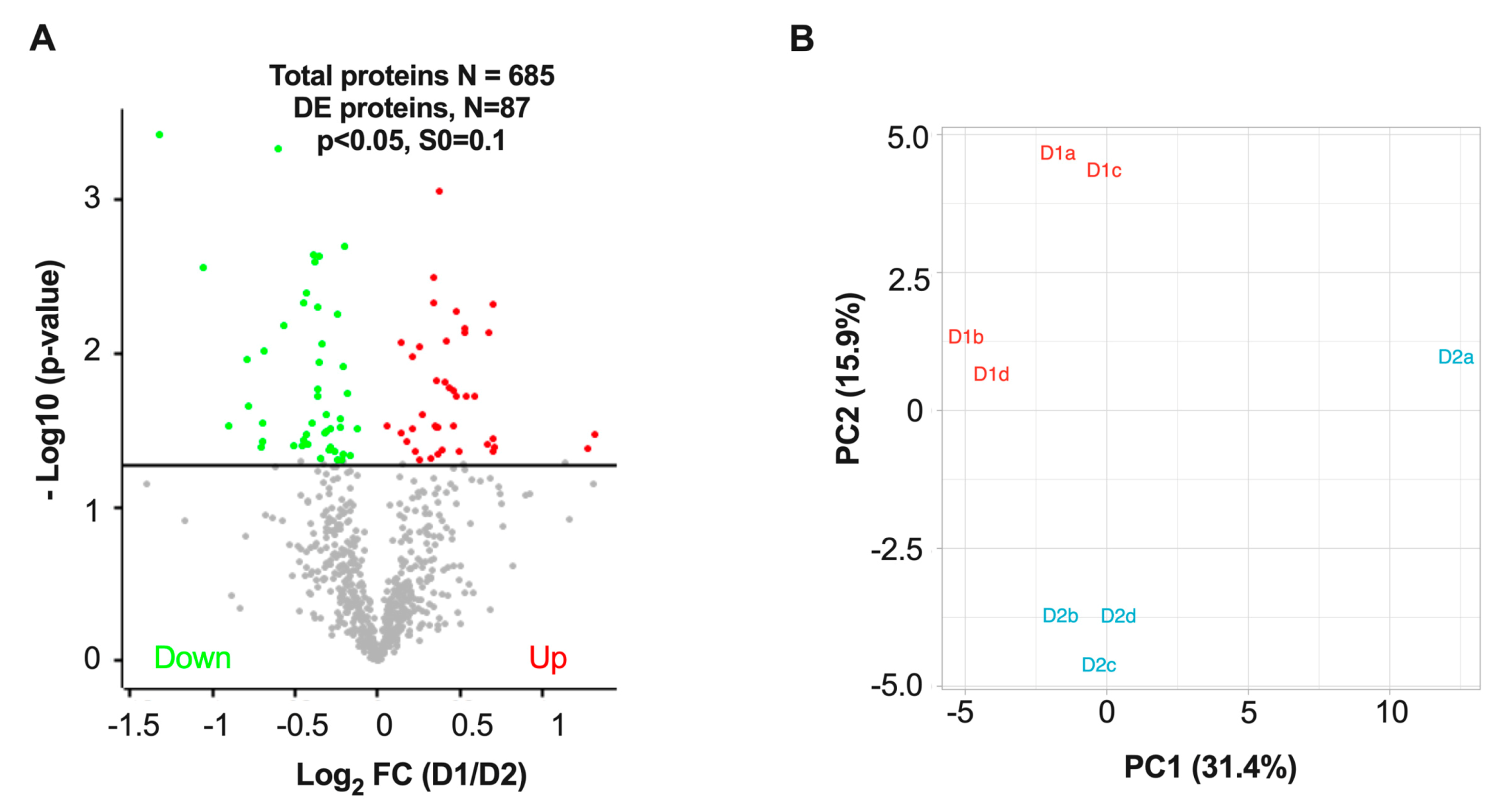
3.3. Differentially Expressed Proteins in D1 and D2 Nuclei Fractions
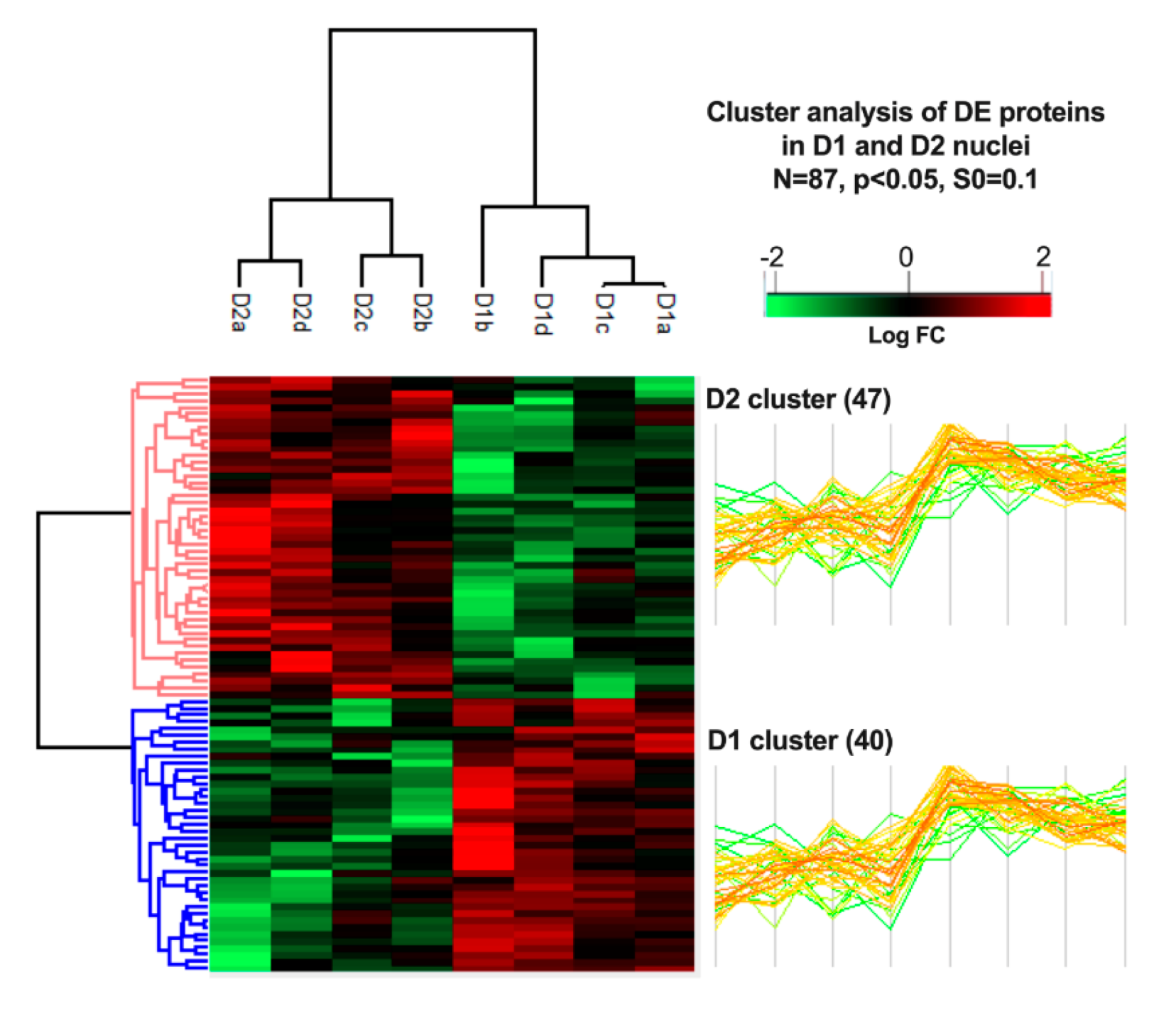
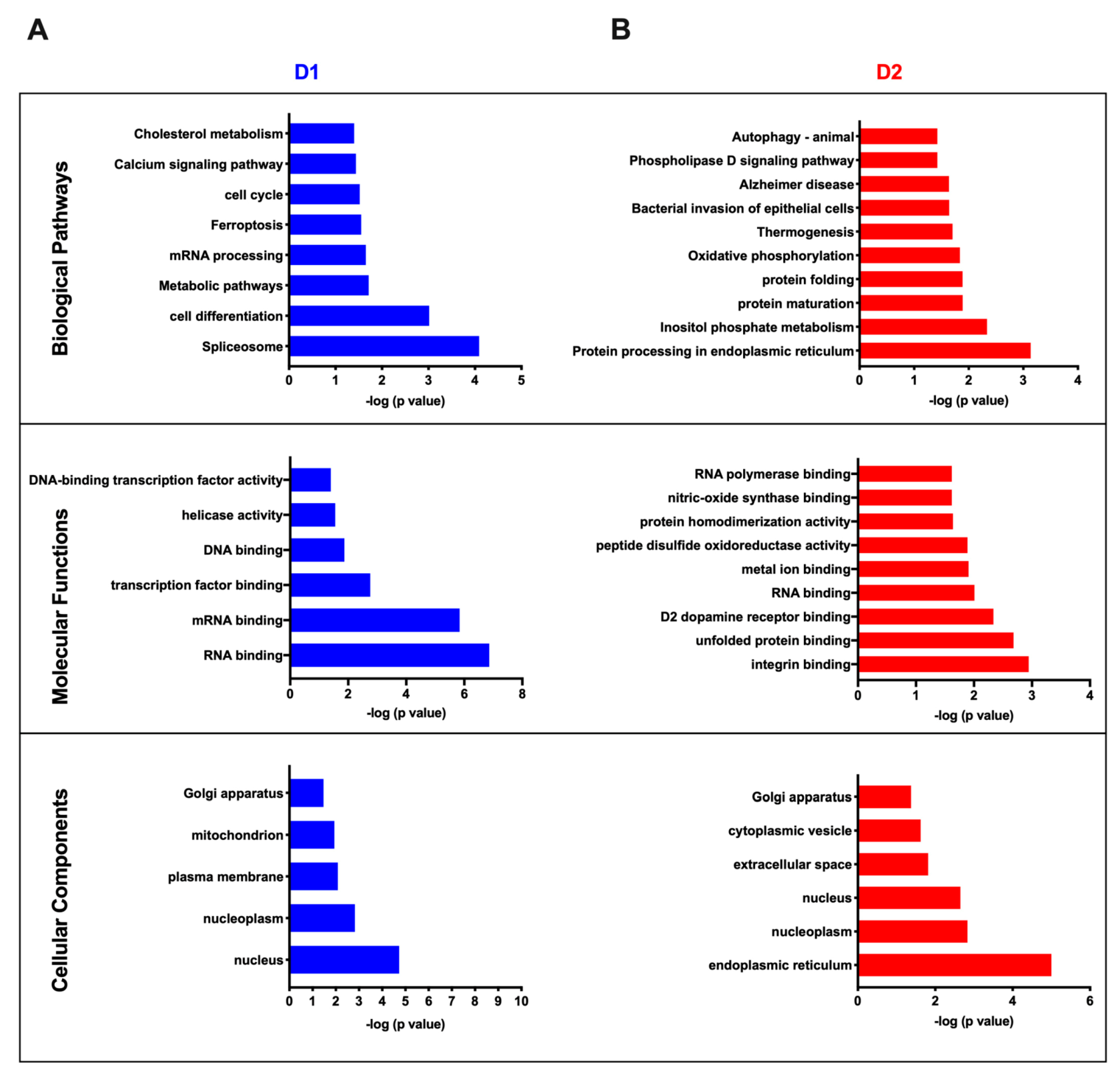
3.4. Cluster Analysis of Differentially Expressed Proteins and Pathway Enrichment
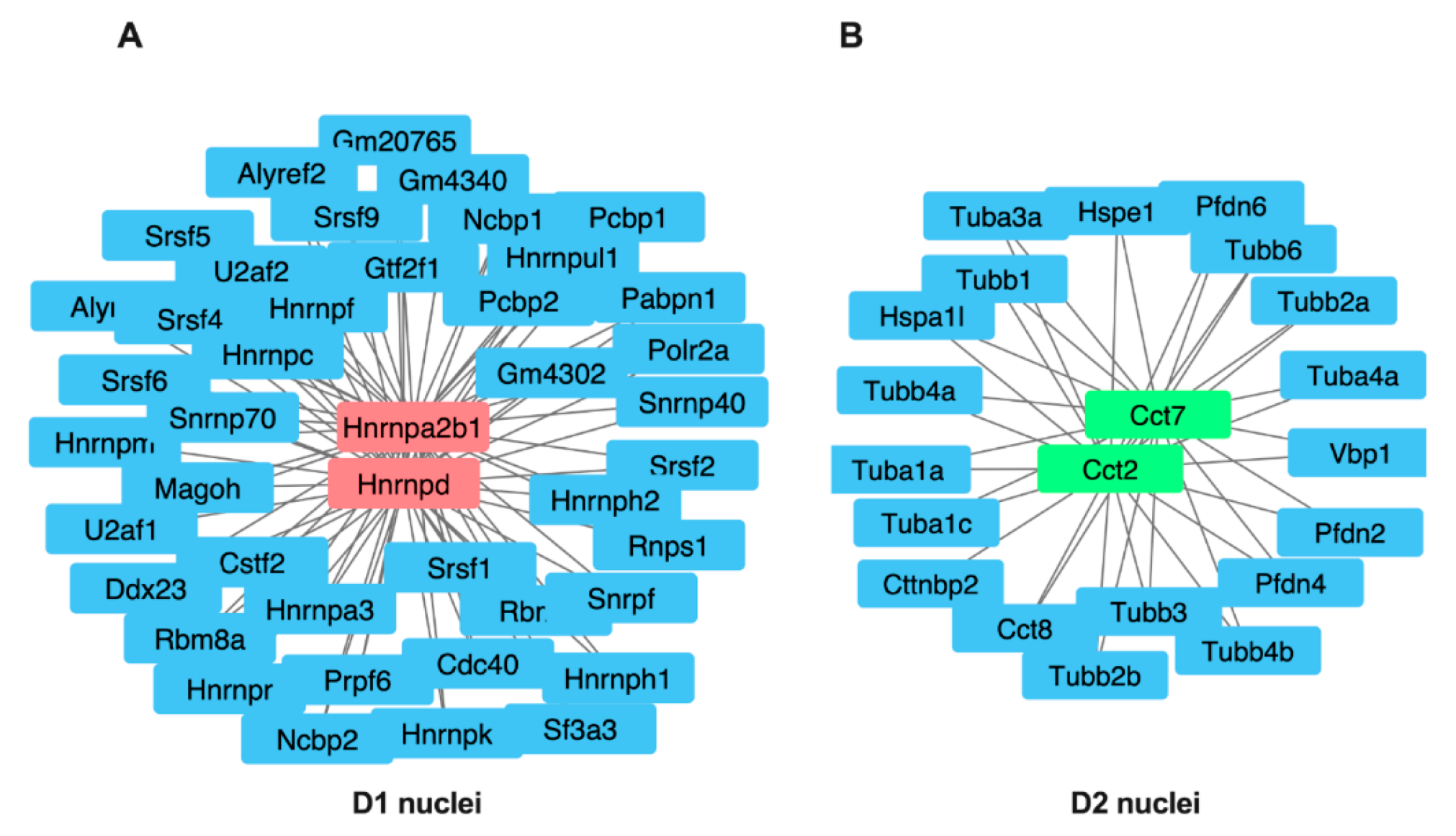
3.5. Central Node Protein Networks in D1 and D2 Cell Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rikani, A.A.; Choudhry, Z.; Choudhry, A.M.; Rizvi, N.; Ikram, H.; Mobassarah, N.J. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Ann. Neurosci. 2014, 21, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Belluscio, M.A.; Escande, M.V.; Keifman, E.; Riquelme, L.A.; Murer, M.G.; Zold, C.L. Oscillations in the basal ganglia in Parkinson’s disease: Role of the striatum. Basal Ganglia 2014, 3, 203–212. [Google Scholar] [CrossRef]
- Greenberg, B.D.; Gabriels, L.A.; Malone, D.A.; Rezai, A.R.; Friehs, G.M.; Okun, M.S.; Shapira, N.A.; Foote, K.D.; Cosyns, P.R.; Kubu, C.S.; et al. Deep brain stimulation of the ventral internal capsule/ventral striatum for obsessive-compulsive disorder: Worldwide experience. Mol. Psychiatry 2008, 15, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.W.; Worhunsky, P.D.; Rogers, R.D.; Goodwin, G.M. Hypoactivation of the ventral and dorsal striatum during reward and loss anticipation in antipsychotic and mood stabilizer-naive bipolar disorder. Neuropsychopharmacology 2014, 40, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Yager, L.M.; Garcia, A.F.; Wunsch, A.M.; Ferguson, S.M. The ins and outs of the striatum: Role in drug addiction. Neuroscience 2015, 301, 529–541. [Google Scholar] [CrossRef]
- Burguière, E.; Monteiro, P.; Mallet, L.; Feng, G.; Graybiel, A.M. Striatal circuits, habits, and implications for obsessive–compulsive disorder. Curr. Opin. Neurobiol. 2015, 30, 59–65. [Google Scholar] [CrossRef]
- Bertran-Gonzalez, J.; Bosch, C.; Maroteaux, M.; Matamales, M.; Hervé, D.; Valjent, E.; Girault, J. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J. Neurosci. 2008, 28, 5671–5685. [Google Scholar] [CrossRef]
- Clark, D.; White, F.J. D1 dopamine receptor—The search for a function: A critical evaluation of the D1/D2 dopamine classification and its functional implications. Synapse 1997, 1, 347–388. [Google Scholar]
- Bateup, H.S.; Svenningsson, P.; Kuroiwa, M.; Gong, S.; Nishi, A.; Heintz, N.; Greengard, P. Cell type–specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci. 2008, 11, 932–939. [Google Scholar] [CrossRef]
- Wang, Y.-Z.; Savas, J.N. Uncovering discrete synaptic proteomes to understand neurological disorders. Proteomes 2018, 6, 30. [Google Scholar] [CrossRef]
- Wilson, R.S.; Nairn, A.C. Cell-type-specific proteomics: A neuroscience perspective. Proteomes 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; De Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; Van Der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular architecture of the mouse nervous system. Cell 2018, 174, 999–1014. [Google Scholar] [CrossRef]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L.; et al. Cell-Type-specific gene expression profiling in adult mouse brain reveals normal and disease-state signatures. Cell Rep. 2019, 26, 2477–2493. [Google Scholar] [CrossRef]
- Rubio, F.J.; Li, X.; Liu, Q.-R.; Cimbro, R.; Hope, B.T. Fluorescence Activated Cell Sorting (FACS) and gene expression analysis of fos-expressing neurons from fresh and frozen rat brain tissue. J. Vis. Exp. 2016, 2016, e54358. [Google Scholar] [CrossRef]
- Berl, S.; Karram, K.; Scheller, A.; Jungblut, M.; Kirchhoff, F.; Waisman, A. Enrichment and isolation of neurons from adult mouse brain for ex vivo analysis. J. Neurosci. Methods 2017, 283, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Xu, J.; Porretta, C.; Nichols, C.D. Neurocytometry: Flow cytometric sorting of specific neuronal populations from human and rodent brain. ACS Chem. Neurosci. 2017, 8, 356–367. [Google Scholar] [CrossRef]
- Jordi, E.; Heiman, M.; Marion-Poll, L.; Guermonprez, P.; Cheng, S.K.; Nairn, A.C.; Greengard, P.; Girault, J. Differential effects of cocaine on histone posttranslational modifications in identified populations of striatal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 9511–9516. [Google Scholar] [CrossRef]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenet. Chromatin 2018, 11, 41. [Google Scholar] [CrossRef]
- Kozlenkov, A.; Wang, M.; Roussos, P.; Rudchenko, S.; Barbu, M.; Bibikova, M.; Klotzle, B.; Dwork, A.J.; Zhang, B.; Hurd, Y.L.; et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 2015, 44, 2593–2612. [Google Scholar] [CrossRef]
- Heiman, M.; Schaefer, A.; Gong, S.; Peterson, J.D.; Day, M.; Ramsey, K.E.; Suárez-Fariñas, M.; Schwarz, C.; Stephan, D.A.; Surmeier, D.J.; et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 2008, 135, 738–748. [Google Scholar] [CrossRef]
- Heiman, M.; Kulicke, R.; Fenster, R.J.; Greengard, P.; Heintz, N. Cell type–specific mRNA purification by translating ribosome affinity purification (TRAP). Nat. Protoc. 2014, 9, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, R.; Kitchen, R.R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Šestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Rhoads, A.; Cocco, A.R.; Peckner, R.; Haber, A.L.; Schweitzer, L.D.; Krug, K.; Mani, D.R.; Clauser, K.R.; Rozenblatt-Rosen, O.; et al. Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells. Mol. Cell. Proteom. 2019, 18, 995–1009. [Google Scholar] [CrossRef]
- Shao, S.; Guo, T.; Gross, V.; Lazarev, A.; Koh, C.C.; Gillessen, S.; Joerger, M.; Jochum, W.; Aebersold, R. Reproducible Tissue homogenization and protein extraction for quantitative proteomics using micropestle-assisted pressure-cycling technology. J. Proteome Res. 2016, 15, 1821–1829. [Google Scholar] [CrossRef]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wühr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Begum, J.; Day, W.; Henderson, C.; Purewal, S.; Cerveira, J.; Summers, H.; Rees, P.; Davies, D.; Filby, A. A method for evaluating the use of fluorescent dyes to track proliferation in cell lines by dye dilution. Cytom. Part A 2013, 83, 1085–1095. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.H.; Woellmer, N.; Koncarevic, S.; Selzer, S.; Böhm, G.; Legner, H.; Schmid, P.; Kienle, S.; Penning, P.; Höhle, C.; et al. TMTpro: Design, synthesis, and initial evaluation of a proline-based isobaric 16-plex tandem mass tag reagent set. Anal. Chem. 2019, 91, 15941–15950. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, K.; Tan, H.; Wu, Z.; Cho, J.-H.; Han, X.; Sun, H.; Beach, T.G.; Peng, J. 27-Plex tandem mass tag mass spectrometry for profiling brain proteome in alzheimer’s disease. Anal. Chem. 2020, 92, 7162–7170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with cytoscape. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef]
- Shao, S.; Guo, T.; Koh, C.C.; Gillessen, S.; Joerger, M.; Jochum, W.; Aebersold, R. Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry. Proteomics 2015, 15, 3711–3721. [Google Scholar] [CrossRef]
- Krishnan, B. Amygdala-hippocampal phospholipase D (PLD) signaling as novel mechanism of cocaine-environment maladaptive conditioned responses. Int. J. Neuropsychopharmacol. 2016, 19, pyv139. [Google Scholar] [CrossRef]
- Gil Oliveira, T.; Di Paolo, G. Phospholipase D in brain function and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 799–805. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Qual. Life Res. 2016, 135, 851–867. [Google Scholar] [CrossRef]
- Chen, X.-Q. Involvement of T-complex protein 1-ring complex/chaperonin containing T-complex protein 1 (TRiC/CCT) in retrograde axonal transport through tau phosphorylation. Neural Regen. Res. 2019, 14, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Dalton, K.; Frydman, J. The mechanism and function of group ii chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Up-Regulated (40) | Down-Regulated (47) | ||||
|---|---|---|---|---|---|
| Gene Name | Fold Change | p-Value | Gene Name | Fold Change | p-Value |
| Tox4 | 2.49116 | 0.0337605 | Coro2a | 0.400584 | 0.00037369 |
| Kctd20 | 2.42119 | 0.0411815 | Hba | 0.481936 | 0.00274324 |
| Hnrnpab | 1.63754 | 0.0409344 | Efhd1 | 0.535936 | 0.02929 |
| Snrpa1 | 1.62469 | 0.0356868 | Septin 11 | 0.578456 | 0.0109418 |
| Me3 | 1.62296 | 0.00481903 | Dnajb11 | 0.583221 | 0.0218579 |
| Eef1e1 | 1.62035 | 0.0429891 | Sfxn5 | 0.611712 | 0.0409574 |
| Elavl1 | 1.59824 | 0.00725727 | Pam | 0.615849 | 0.037189 |
| Nup133 | 1.58732 | 0.0387323 | Dlg4 | 0.616571 | 0.0285791 |
| Vdac3 | 1.50586 | 0.0188459 | Sacm1l | 0.619851 | 0.00970264 |
| Lsm6 | 1.45014 | 0.0190832 | Aldoc | 0.658441 | 0.00046328 |
| Zfp292 | 1.43915 | 0.00725454 | Ncam1 | 0.676135 | 0.00650359 |
| Hnrnpa1 | 1.43844 | 0.00683687 | Sfxn3 | 0.703261 | 0.0397627 |
| D1Pas1 | 1.40947 | 0.0434996 | Rps11 | 0.726778 | 0.0394326 |
| Eftud2 | 1.39301 | 0.00532517 | Cct2 | 0.732122 | 0.0366409 |
| Hnrnpa0 | 1.3922 | 0.0188692 | Pabpn1 | 0.733746 | 0.0046371 |
| Celf2 | 1.37592 | 0.0292762 | Pgap1 | 0.740795 | 0.00399328 |
| Hnrnpdl | 1.37127 | 0.0172432 | Gabarapl2 | 0.742762 | 0.0333492 |
| Syncrip | 1.35272 | 0.0166647 | Dctn3 | 0.74543 | 0.0392584 |
| Rbmx | 1.33812 | 0.00827693 | Rpl24 | 0.761663 | 0.0281739 |
| Purb | 1.32497 | 0.0154027 | Cct7 | 0.763964 | 0.00226032 |
| Pura | 1.30983 | 0.0424454 | Slc4a4 | 0.768653 | 0.00254452 |
| Hnrnpd | 1.29834 | 0.00088056 | Ppib | 0.777091 | 0.0189307 |
| Npm1 | 1.29117 | 0.0299636 | Slc25a3 | 0.777111 | 0.00503528 |
| Ptbp2 | 1.29086 | 0.0454053 | Pdia6 | 0.778812 | 0.0170188 |
| Tomm70 | 1.28277 | 0.0149869 | Slc25a5 | 0.780524 | 0.0112892 |
| Hnrnpl | 1.27938 | 0.0301104 | Vps35 | 0.780705 | 0.00233104 |
| Ddx17 | 1.2712 | 0.0293465 | Sec61a1 | 0.788051 | 0.0480851 |
| Eif4a3 | 1.26848 | 0.00472075 | Cct8 | 0.789875 | 0.00857058 |
| Gnal | 1.26502 | 0.00318639 | Apmap | 0.800946 | 0.0324994 |
| Atp6v1b2 | 1.24752 | 0.0477972 | Slc3a2 | 0.805497 | 0.0251531 |
| Srrm2 | 1.21029 | 0.0249769 | Tmpo | 0.807824 | 0.0318018 |
| Rbmxl1 | 1.19467 | 0.00896132 | Asrgl1 | 0.815023 | 0.0427078 |
| Hnrnpu | 1.19465 | 0.0490688 | Dctn2 | 0.819532 | 0.0305211 |
| Erh | 1.17264 | 0.0428214 | Ganab | 0.820572 | 0.0307506 |
| Ruvbl1 | 1.16093 | 0.0104841 | Plch1 | 0.821746 | 0.0407058 |
| Hnrnpa2b1 | 1.15681 | 0.0310403 | Ckap4 | 0.835119 | 0.0435046 |
| Ndufa6 | 1.13342 | 0.0371455 | Dnm2 | 0.845664 | 0.0485707 |
| Prpf19 | 1.10611 | 0.0325835 | Calr | 0.847391 | 0.0055328 |
| Cplx1 | 1.10327 | 0.00840265 | Sv2a | 0.854525 | 0.0302238 |
| Vdac2 | 1.04319 | 0.0298126 | Glud1 | 0.857564 | 0.0264393 |
| Hadha | 0.858226 | 0.0489431 | |||
| P4hb | 0.864091 | 0.0451958 | |||
| Thy1 | 0.866795 | 0.012009 | |||
| Rab6a | 0.870134 | 0.00202572 | |||
| Ctsd | 0.882804 | 0.0180612 | |||
| Dnm1 | 0.889323 | 0.0456554 | |||
| Hpcal4 | 0.919234 | 0.0307775 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansuri, M.S.; Peng, G.; Wilson, R.S.; Lam, T.T.; Zhao, H.; Williams, K.R.; Nairn, A.C. Differential Protein Expression in Striatal D1- and D2-Dopamine Receptor-Expressing Medium Spiny Neurons. Proteomes 2020, 8, 27. https://doi.org/10.3390/proteomes8040027
Mansuri MS, Peng G, Wilson RS, Lam TT, Zhao H, Williams KR, Nairn AC. Differential Protein Expression in Striatal D1- and D2-Dopamine Receptor-Expressing Medium Spiny Neurons. Proteomes. 2020; 8(4):27. https://doi.org/10.3390/proteomes8040027
Chicago/Turabian StyleMansuri, M. Shahid, Gang Peng, Rashaun S. Wilson, TuKiet T. Lam, Hongyu Zhao, Kenneth R. Williams, and Angus C. Nairn. 2020. "Differential Protein Expression in Striatal D1- and D2-Dopamine Receptor-Expressing Medium Spiny Neurons" Proteomes 8, no. 4: 27. https://doi.org/10.3390/proteomes8040027
APA StyleMansuri, M. S., Peng, G., Wilson, R. S., Lam, T. T., Zhao, H., Williams, K. R., & Nairn, A. C. (2020). Differential Protein Expression in Striatal D1- and D2-Dopamine Receptor-Expressing Medium Spiny Neurons. Proteomes, 8(4), 27. https://doi.org/10.3390/proteomes8040027