Targeting a Subset of the Membrane Proteome for Top–Down Mass Spectrometry: Introducing the Proteolipidome


Abstract
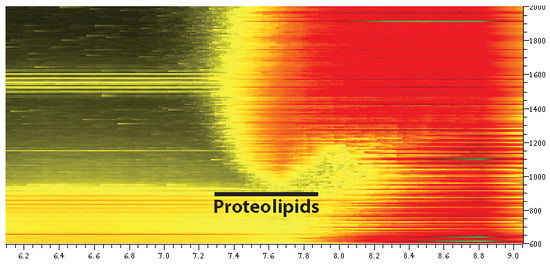
{kind=link}
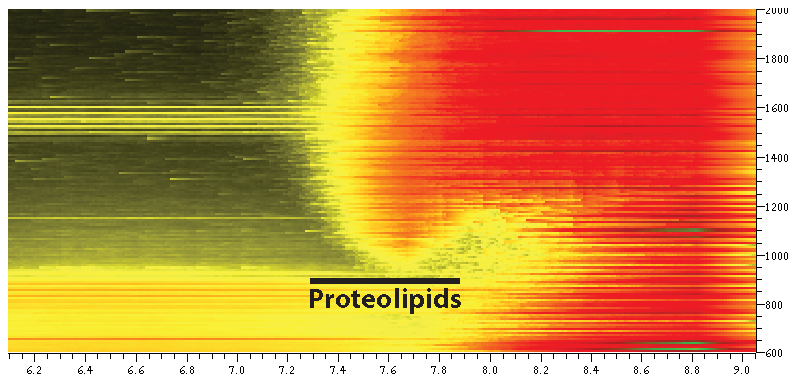
{kind=link}
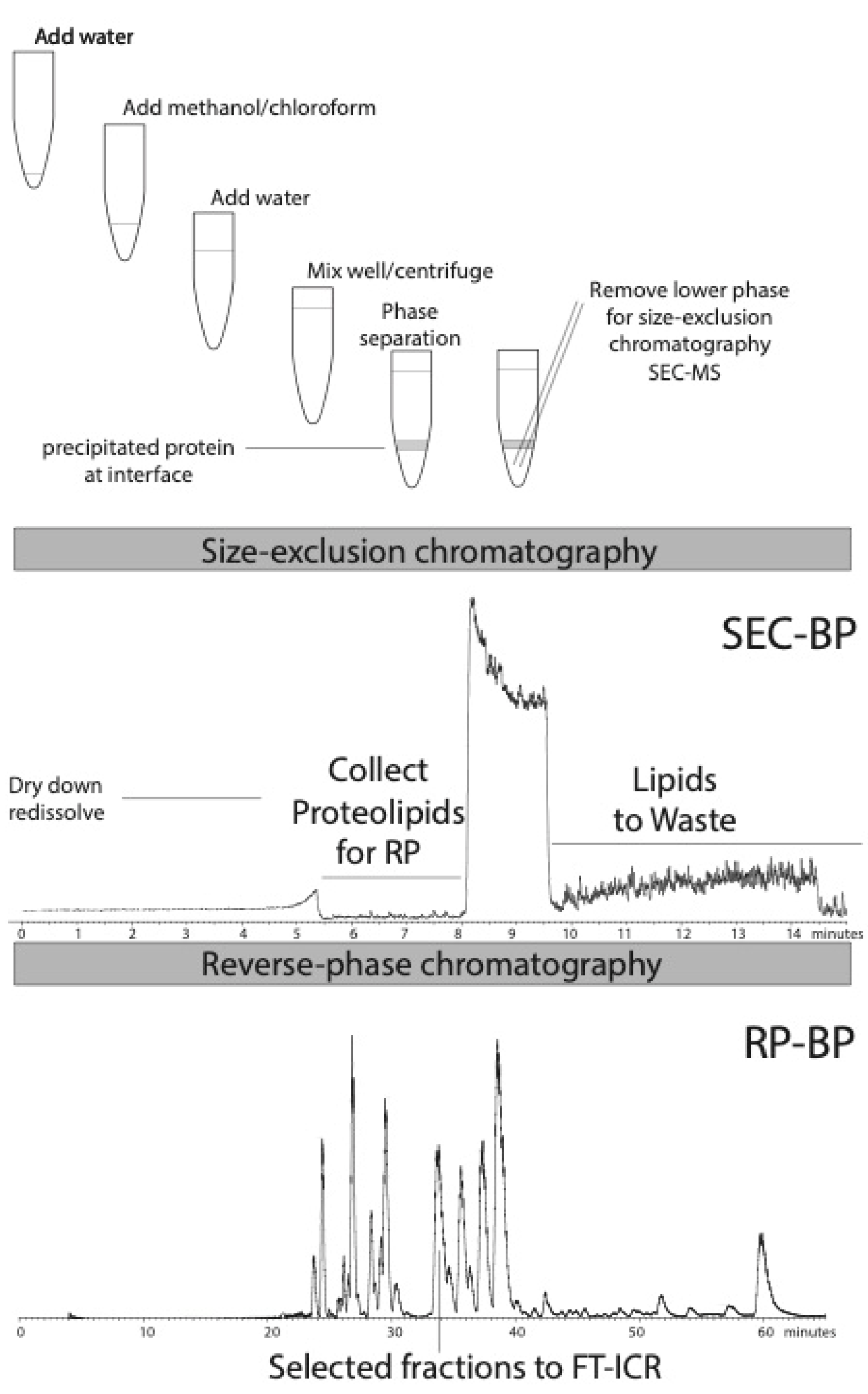
{kind=link}
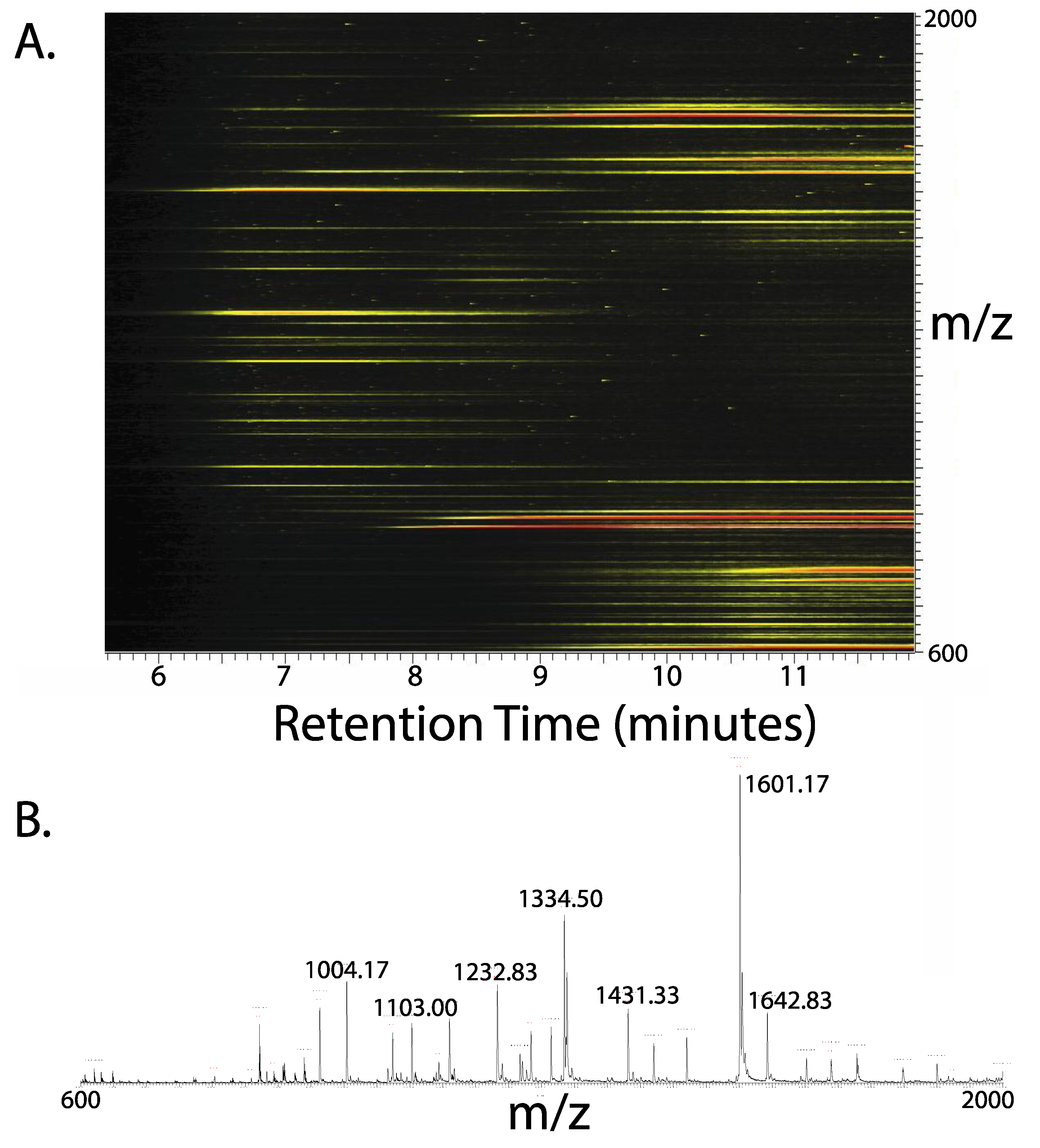
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
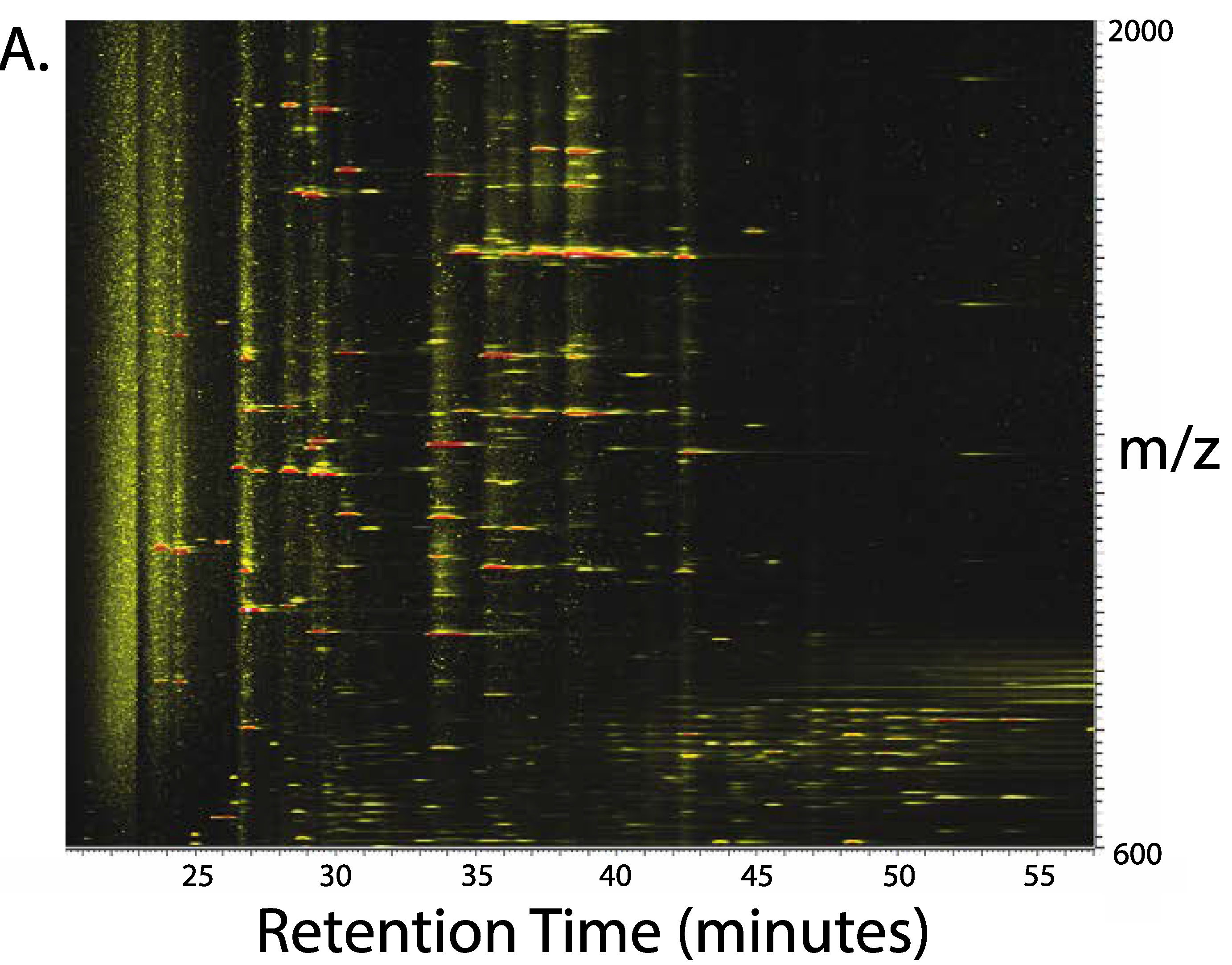
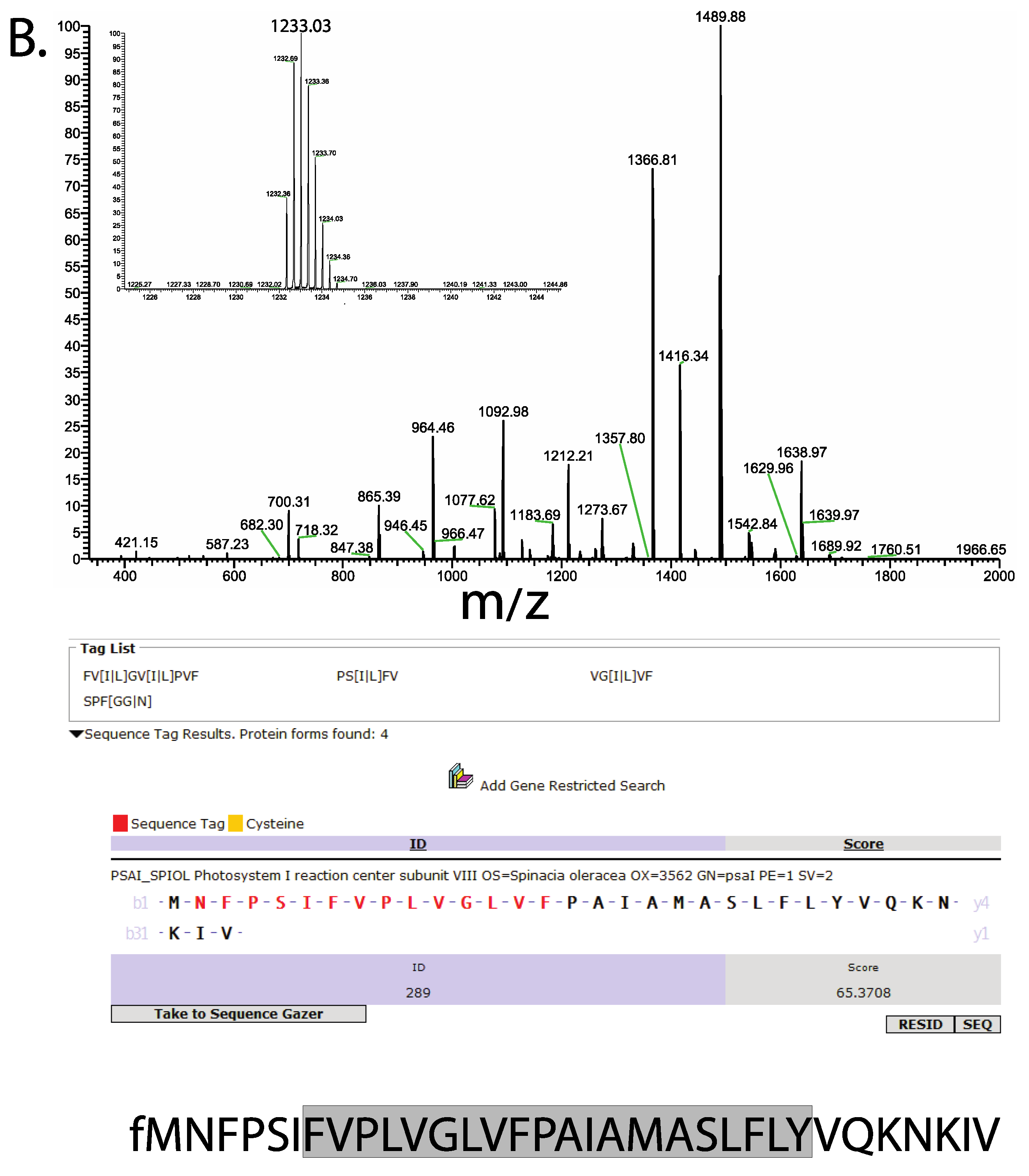
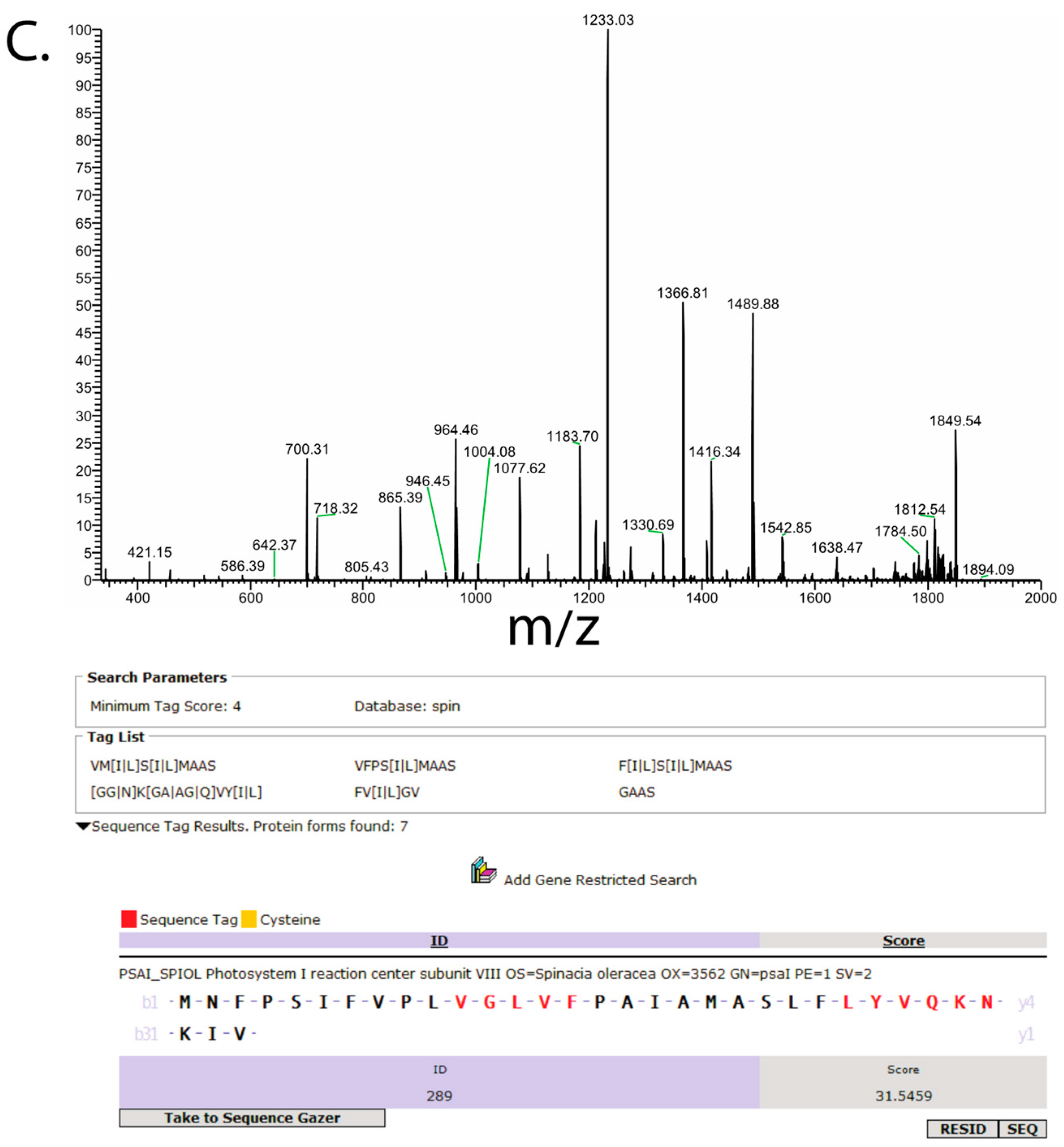
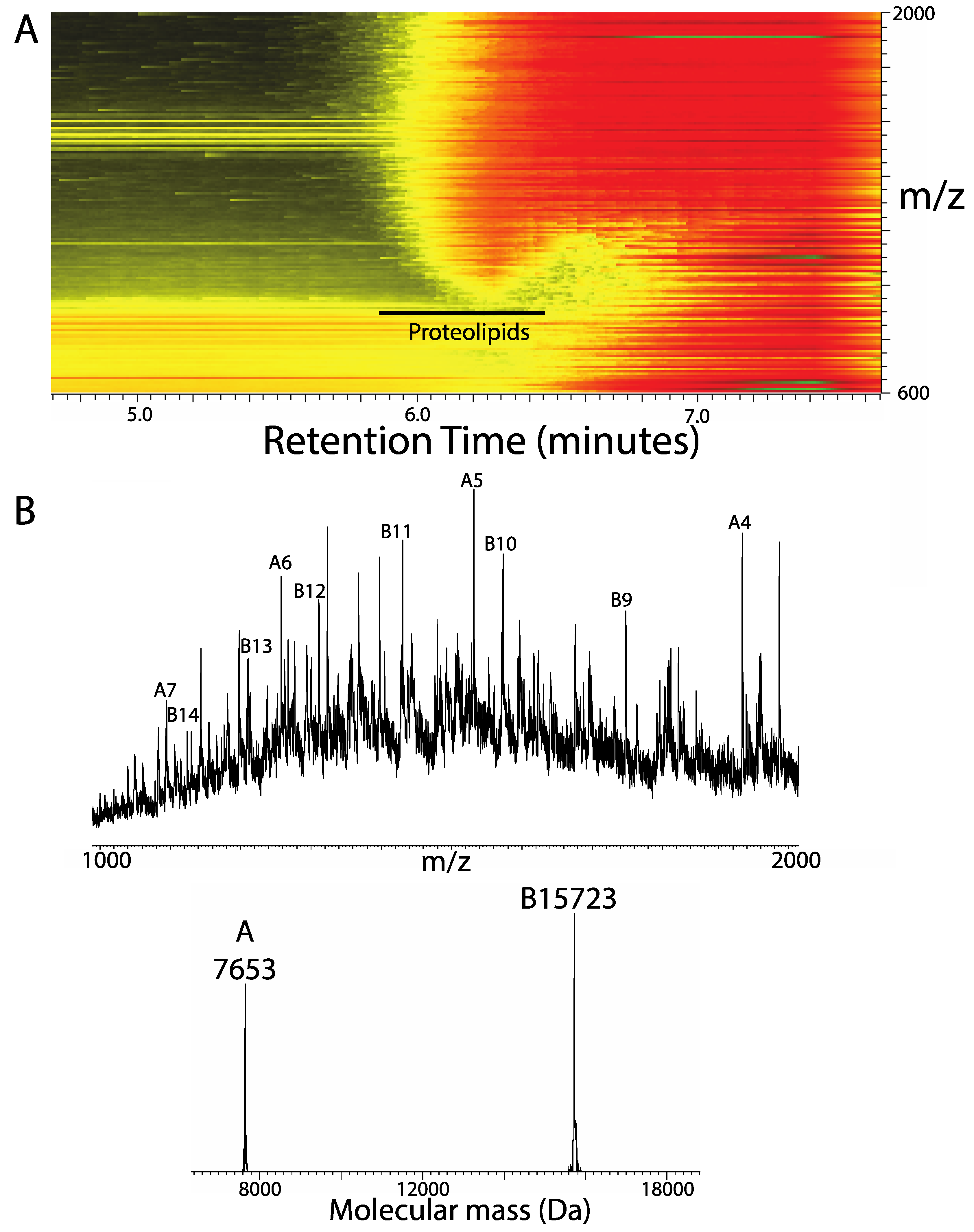
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Santoni, V.; Molloy, M.; Rabilloud, T. Membrane proteins and proteomics: Un amour impossible? Electrophoresis 2000, 21, 1054–1070. [Google Scholar] [CrossRef]
- Blonder, J.; Conrads, T.P.; Yu, L.R.; Terunuma, A.; Janini, G.M.; Issaq, H.J.; Vogel, J.C.; Veenstra, T.D. A detergent- and cyanogen bromide-free method for integral membrane proteomics: Application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics 2004, 4, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; MacCoss, M.J.; Howell, K.E.; Yates, J.R., 3rd. A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 2003, 21, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Speers, A.E.; Blackler, A.R.; Wu, C.C. Shotgun analysis of integral membrane proteins facilitated by elevated temperature. Anal. Chem. 2007, 79, 4613–4620. [Google Scholar] [CrossRef] [PubMed]
- Whitelegge, J.P.; Jewess, P.; Pickering, M.G.; Gerrish, C.; Camilleri, P.; Bowyer, J.R. Sequence analysis of photoaffinity-labelled peptides derived by proteolysis of photosystem-2 reaction centres from thylakoid membranes treated with [14C]azidoatrazine. Eur. J. Biochem. 1992, 207, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Szule, J.A.; Jarvis, S.E.; Hibbert, J.E.; Spafford, J.D.; Braun, J.E.; Zamponi, G.W.; Wessel, G.M.; Coorssen, J.R. Calcium-triggered membrane fusion proceeds independently of specific presynaptic proteins. J. Biol. Chem. 2003, 278, 24251–24254. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Wolters, D.; Rogner, M.; Poetsch, A. Toward the complete membrane proteome: High coverage of integral membrane proteins through transmembrane peptide detection. Mol. Cell. Proteomics 2006, 5, 444–453. [Google Scholar] [CrossRef]
- Whitelegge, J.P.; Zhang, H.; Aguilera, R.; Taylor, R.M.; Cramer, W.A. Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein: Cytochrome b(6)f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol. Cell. Proteomics 2002, 1, 816–827. [Google Scholar] [CrossRef]
- Whitelegge, J.P. Integral membrane proteins and bilayer proteomics. Anal. Chem. 2013, 85, 2558–2568. [Google Scholar] [CrossRef]
- Robinson, C.V. Mass spectrometry: From plasma proteins to mitochondrial membranes. Proc. Natl. Acad. Sci. USA 2019, 116, 2814–2820. [Google Scholar] [CrossRef] [PubMed]
- Chatagnon, C.; Mortreuil, M.; Zalta, J.P.; Chatagnon, P. Analysis of proteolipid proteins of human and bovine brains. Bull. Soc. Chim. Biol. (Paris) 1953, 35, 419–422. [Google Scholar]
- Tzagoloff, A.; Akai, A.; Foury, F. Assembly of the mitochondrial membrane system XVI. Modified form of the ATPase proteolipid in oligomycin-resistant mutants of Saccharomyces cerevisiae. FEBS Lett. 1976, 65, 391–395. [Google Scholar] [CrossRef]
- Girvin, M.E.; Fillingame, R.H. Helical structure and folding of subunit c of F1F0 ATP synthase: 1H NMR resonance assignments and NOE analysis. Biochemistry 1993, 32, 12167–12177. [Google Scholar] [CrossRef] [PubMed]
- Zabrouskov, V.; Whitelegge, J.P. Increased coverage in the transmembrane domain with activated-ion electron capture dissociation for top-down Fourier-transform mass spectrometry of integral membrane proteins. J. Proteome Res. 2007, 6, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Whitelegge, J.P.; le Coutre, J.; Lee, J.C.; Engel, C.K.; Prive, G.G.; Faull, K.F.; Kaback, H.R. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact transmembrane proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 10695–10698. [Google Scholar] [CrossRef]
- le Coutre, J.; Whitelegge, J.P.; Gross, A.; Turk, E.; Wright, E.M.; Kaback, H.R.; Faull, K.F. Proteomics on full-length membrane proteins using mass spectrometry. Biochemistry 2000, 39, 4237–4242. [Google Scholar] [CrossRef] [PubMed]
- Whitelegge, J.P.; Gundersen, C.B.; Faull, K.F. Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci. 1998, 7, 1423–1430. [Google Scholar] [CrossRef]
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Thangaraj, B.; Ryan, C.M.; Souda, P.; Krause, K.; Faull, K.F.; Weber, A.P.; Fromme, P.; Whitelegge, J.P. Data-directed top-down Fourier-transform mass spectrometry of a large integral membrane protein complex: Photosystem II from Galdieria sulphuraria. Proteomics 2010, 10, 3644–3656. [Google Scholar] [CrossRef]
- Gomez, S.M.; Nishio, J.N.; Faull, K.F.; Whitelegge, J.P. The chloroplast grana proteome defined by intact mass measurements from liquid chromatography mass spectrometry. Mol. Cell. Proteomics 2002, 1, 46–59. [Google Scholar] [CrossRef]
- Whitelegge, J.P. Mass spectrometry for high throughput quantitative proteomics in plant research: Lessons from thylakoid membranes. Plant. Physiol. Biochem. 2004, 42, 919–927. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Yergey, A.L. Proteomics Is Analytical Chemistry: Fitness-for-Purpose in the Application of Top-Down and Bottom-Up Analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef]
- Whitelegge, J.P. Plant proteomics: BLASTing out of a MudPIT. Proc. Natl. Acad. Sci. USA 2002, 99, 11564–11566. [Google Scholar] [CrossRef]
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Fornelli, L.; Toby, T.K.; Schachner, L.F.; Doubleday, P.F.; Srzentic, K.; DeHart, C.J.; Kelleher, N.L. Top-down proteomics: Where we are, where we are going? J. Proteomics 2018, 175, 3–4. [Google Scholar] [CrossRef]
- Laganowsky, A.; Reading, E.; Allison, T.M.; Ulmschneider, M.B.; Degiacomi, M.T.; Baldwin, A.J.; Robinson, C.V. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 2014, 510, 172–175. [Google Scholar] [CrossRef]
- Chorev, D.S.; Baker, L.A.; Wu, D.; Beilsten-Edmands, V.; Rouse, S.L.; Zeev-Ben-Mordehai, T.; Jiko, C.; Samsudin, F.; Gerle, C.; Khalid, S.; et al. Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry. Science 2018, 362, 829–834. [Google Scholar] [CrossRef]
- Hirst, J.; Kunji, E.R.S.; Walker, J.E. Comment on “Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry”. Science 2019, 366. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitelegge, J. Targeting a Subset of the Membrane Proteome for Top–Down Mass Spectrometry: Introducing the Proteolipidome. Proteomes 2020, 8, 5. https://doi.org/10.3390/proteomes8010005
Whitelegge J. Targeting a Subset of the Membrane Proteome for Top–Down Mass Spectrometry: Introducing the Proteolipidome. Proteomes. 2020; 8(1):5. https://doi.org/10.3390/proteomes8010005
Chicago/Turabian StyleWhitelegge, Julian. 2020. "Targeting a Subset of the Membrane Proteome for Top–Down Mass Spectrometry: Introducing the Proteolipidome" Proteomes 8, no. 1: 5. https://doi.org/10.3390/proteomes8010005
APA StyleWhitelegge, J. (2020). Targeting a Subset of the Membrane Proteome for Top–Down Mass Spectrometry: Introducing the Proteolipidome. Proteomes, 8(1), 5. https://doi.org/10.3390/proteomes8010005