Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity

Abstract
:1. Introduction
2. AMPAR Complex
3. Phosphorylation of the Pore-Forming Subunits: GluA1
4. Significance of GluA1 Phosphorylation for Synaptic Plasticity: Knock-In Mouse Studies
5. Phosphorylation of the Pore-Forming Subunits: GluA2 and GluA3
6. Phosphorylation of the Pore-Forming Subunits: GluA4
7. TARP Phosphorylation and Its Roles in LTP
8. Phosphorylation of Other Auxiliary Proteins of AMPAR
9. Closing Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Malenka, R.C.; Kauer, J.A.; Perkel, D.J.; Mauk, M.D.; Kelly, P.T.; Nicoll, R.A.; Waxham, M.N. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 1989, 340, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Malinow, R.; Schulman, H.; Tsien, R.W. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 1989, 245, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Stevens, C.F.; Tonegawa, S.; Wang, Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 1992, 257, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A.; Tomita, S.; Bredt, D.S. Auxiliary subunits assist AMPA-type glutamate receptors. Science 2006, 311, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Nicoll, R.A. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 2011, 70, 178–199. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Yelshanskaya, M.V.; Grassucci, R.A.; Frank, J.; Sobolevsky, A.I. Elucidation of AMPA receptor-stargazin complexes by cryo-electron microscopy. Science 2016, 353, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhao, Y.; Wang, Y.; Shekhar, M.; Tajkhorshid, E.; Gouaux, E. Activation and desensitization mechanism of AMPA receptor-TARP complex by cryo-EM. Cell 2017, 170, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Tomita, S. Defined criteria for auxiliary subunits of glutamate receptors. J. Physiol. 2012, 590, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Zamanillo, D.; Sprengel, R.; Hvalby, O.; Jensen, V.; Burnashev, N.; Rozov, A.; Kaiser, K.M.; Köster, H.J.; Borchardt, T.; Worley, P.; et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 1999, 284, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Byrd, K.; Petralia, R.S.; Elias, G.M.; Adesnik, H.; Tomita, S.; Karimzadegan, S.; Kealey, C.; Bredt, D.S.; Nicoll, R.A. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat. Neurosci. 2005, 8, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.E.; Shi, Y.; Suh, Y.H.; Zheng, C.Y.; Blankenship, S.M.; Roche, K.W.; Nicoll, R.A. Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron 2013, 77, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Steinberg, J.P.; Huganir, R.L.; Linden, D.J. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 2003, 300, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Wu, L.J.; Zhao, M.G.; Xu, H.; Jia, Z.; Zhuo, M. Long-term depression requires postsynaptic AMPA GluR2 receptor in adult mouse cingulate cortex. J. Cell. Physiol. 2007, 211, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Arora, A.; Yang, L.; Parelkar, N.K.; Zhang, G.; Liu, X.; Choe, E.S.; Mao, L. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol. Neurobiol. 2005, 32, 237–249. [Google Scholar] [CrossRef]
- Hollmann, M.; Maron, C.; Heinemann, S. N-Glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron 1994, 13, 1331–1343. [Google Scholar] [CrossRef]
- Barria, A.; Derkach, V.; Soderling, T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J. Biol. Chem. 1997, 272, 32727–32730. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.W.; O’Brien, R.J.; Mammen, A.L.; Bernhardt, J.; Huganir, R.L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 1996, 16, 1179–1188. [Google Scholar] [CrossRef]
- Mammen, A.L.; Kameyama, K.; Roche, K.W.; Huganir, R.L. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J. Biol. Chem. 1997, 272, 32528–32533. [Google Scholar] [CrossRef] [PubMed]
- Derkach, V.; Barria, A.; Soderling, T.R. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 3269–3274. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Traynelis, S.F. PKC phosphorylates GluA1-Ser831 to enhance AMPA receptor conductance. Channels 2012, 6, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Banke, T.G.; Bowie, D.; Lee, H.K.; Huganir, R.L.; Schousboe, A.; Traynelis, S.F. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 2000, 20, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Barbarosie, M.; Kameyama, K.; Bear, M.F.; Huganir, R.L. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 2000, 405, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.P.; Fedorov, N.B.; Filipkowski, R.K.; Silva, A.J. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.; Kang, M.G.; Johnson, R.C.; Esteban, J.; Huganir, R.L.; Malinow, R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 2006, 51, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Takamiya, K.; Kameyama, K.; He, K.; Yu, S.; Rossetti, L.; Wilen, D.; Huganir, R.L. Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol. Cell. Neurosci. 2007, 36, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, E.E.; Guglietta, R.; Khakh, B.S.; O’Dell, T.J. Inhibitory interactions between phosphorylation sites in the C terminus of α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor GluA1 subunits. J. Biol. Chem. 2014, 289, 14600–14611. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Wells, G.; Bachman, J.; Snyder, J.P.; Jenkins, A.; Huganir, R.L.; Oswald, R.E.; Traynelis, S.F. Regulation of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol. Pharmacol. 2014, 85, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N.K.; Thomas, G.M.; Luo, J.; Huganir, R.L. Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E5883–E5890. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Isozaki, K.; Roche, K.W.; Nicoll, R.A. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc. Natl. Acad. Sci. USA 2010, 107, 22266–22271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.J.; Chung, H.J.; Huganir, R.L. Identification of protein kinase C phosphorylation sites within the AMPA receptor GluR2 subunit. Neuropharmacology 2001, 41, 672–679. [Google Scholar] [CrossRef]
- Hayashi, T.; Huganir, R.L. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 2004, 24, 6152–6160. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Mikawa, S.; Hirai, H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J. Neurochem. 1999, 73, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, G.; Ju, W.; Liu, L.; Wyszynski, M.; Lee, S.H.; Dunah, A.W.; Taghibiglou, C.; Wang, Y.; Lu, J.; Wong, T.P.; et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004, 23, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, A.L.; Kameyama, K.; Huganir, R.L. Characterization of phosphorylation sites on the glutamate receptor 4 subunit of the AMPA receptors. J. Neurosci. 1999, 19, 4748–4754. [Google Scholar] [CrossRef] [PubMed]
- Esteban, J.A.; Shi, S.H.; Wilson, C.; Nuriya, M.; Huganir, R.L.; Malinow, R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 2003, 6, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Stein, V.; Stocker, T.J.; Nicoll, R.A.; Bredt, D.S. Bidirectional synaptic plasticity regulated by phosphorylation of stargazing-like TARPs. Neuron 2005, 45, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, A.; Yan, D.; Tomita, S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 2010, 66, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.S.; Penn, A.C.; Grillo-Bosch, D.; Retailleau, N.; Poujol, C.; Philippat, A.; Coussen, F.; Sainlos, M.; Opazo, P.; Choquet, D. Lengthening of the Stargazin cytoplasmic tail increases synaptic transmission by promoting interaction to deeper domains of PSD-95. Neuron 2015, 86, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.L.; Chetkovich, D.M. Regulation of stargazin synaptic trafficking by C-terminal PDZ ligand phosphorylation in bidirectional synaptic plasticity. J. Neurochem. 2010, 113, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Chávez, A.E.; Mineur, Y.S.; Morimoto-Tomita, M.; Lutzu, S.; Kim, K.S.; Picciotto, M.R.; Castillo, P.E.; Tomita, S. CaMKII phosphorylation of TARPγ-8 is a mediator of LTP and learning and memory. Neuron 2016, 92, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Kunde, S.A.; Rademacher, N.; Zieger, H.; Shoichet, S.A. Protein kinase C regulates AMPA receptor auxiliary protein Shisa9/CKAMP44 through interactions with neuronal scaffold PICK1. FEBS Open Bio 2017, 7, 1234–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Takamiya, K.; Han, J.S.; Man, H.; Kim, C.H.; Rumbaugh, G.; Yu, S.; Ding, L.; He, C.; Petralia, R.S.; et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 2003, 112, 631–643. [Google Scholar] [CrossRef]
- Lee, H.K.; Takamiya, K.; He, K.; Song, L.; Huganir, R.L. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 2010, 103, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; O’Brien, R.J.; Fung, E.T.; Lanahan, A.A.; Worley, P.F.; Huganir, R.L. GRIP: A synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 1997, 386, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tigaret, C.; Choquet, D. More AMPAR garnish. Science 2009, 323, 1295–1296. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Cull-Candy, S.G. Neuroscience. AMPA receptors—Another twist? Science 2010, 327, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Haering, S.C.; Tapken, D.; Pahl, S.; Hollmann, M. Auxiliary subunits: shepherding AMPA receptors to the plasma membrane. Membranes 2014, 4, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Fukaya, M.; Qiao, X.; Sakimura, K.; Watanabe, M.; Kano, M. Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J. Neurosci. 1999, 19, 6027–6036. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chetkovich, D.M.; Petralia, R.S.; Sweeney, N.T.; Kawasaki, Y.; Wenthold, R.J.; Bredt, D.S.; Nicoll, R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Chen, L.; Kawasaki, Y.; Petralia, R.S.; Wenthold, R.J.; Nicoll, R.A.; Bredt, D.S. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell Biol. 2003, 161, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, S.; Adesnik, H.; Sekiguchi, M.; Zhang, W.; Wada, K.; Howe, J.R.; Nicoll, R.A.; Bredt, D.S. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 2005, 435, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Zhou, W.; Milstein, A.D.; Knierman, M.D.; Siuda, E.R.; Dotzlaf, J.E.; Yu, H.; Hale, J.E.; Nisenbaum, E.S.; Nicoll, R.A.; et al. New transmembrane AMPA receptor regulatory protein isoform, gamma-7, differentially regulates AMPA receptors. J. Neurosci. 2007, 27, 4969–4977. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Siuda, E.R.; Nisenbaum, E.S.; Bredt, D.S. AMPA receptor subunit-specific regulation by a distinct family of type II TARPs. Neuron 2008, 59, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, A.; Brown, T.E.; Kato, A.S.; Bredt, D.S.; Kauer, J.A.; Tomita, S. PDZ binding of TARPγ-8 controls synaptic transmission but not synaptic plasticity. Nat. Neurosci. 2011, 14, 1410–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenk, J.; Harmel, N.; Zolles, G.; Bildl, W.; Kulik, A.; Heimrich, B.; Chisaka, O.; Jonas, P.; Schulte, U.; Fakler, B.; et al. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 2009, 323, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.S.; Gill, M.B.; Ho, M.T.; Yu, H.; Tu, Y.; Siuda, E.R.; Wang, H.; Qian, Y.W.; Nisenbaum, E.S.; Tomita, S.; et al. Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron 2010, 68, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Harmel, N.; Brechet, A.; Zolles, G.; Berkefeld, H.; Müller, C.S.; Bildl, W.; Baehrens, D.; Hüber, B.; Kulik, A.; et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 2012, 74, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Shanks, N.F.; Savas, J.N.; Maruo, T.; Cais, O.; Hirao, A.; Oe, S.; Ghosh, A.; Noda, Y.; Greger, I.H.; Yates, J.R., III; et al. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012, 1, 590–598. [Google Scholar] [CrossRef] [PubMed]
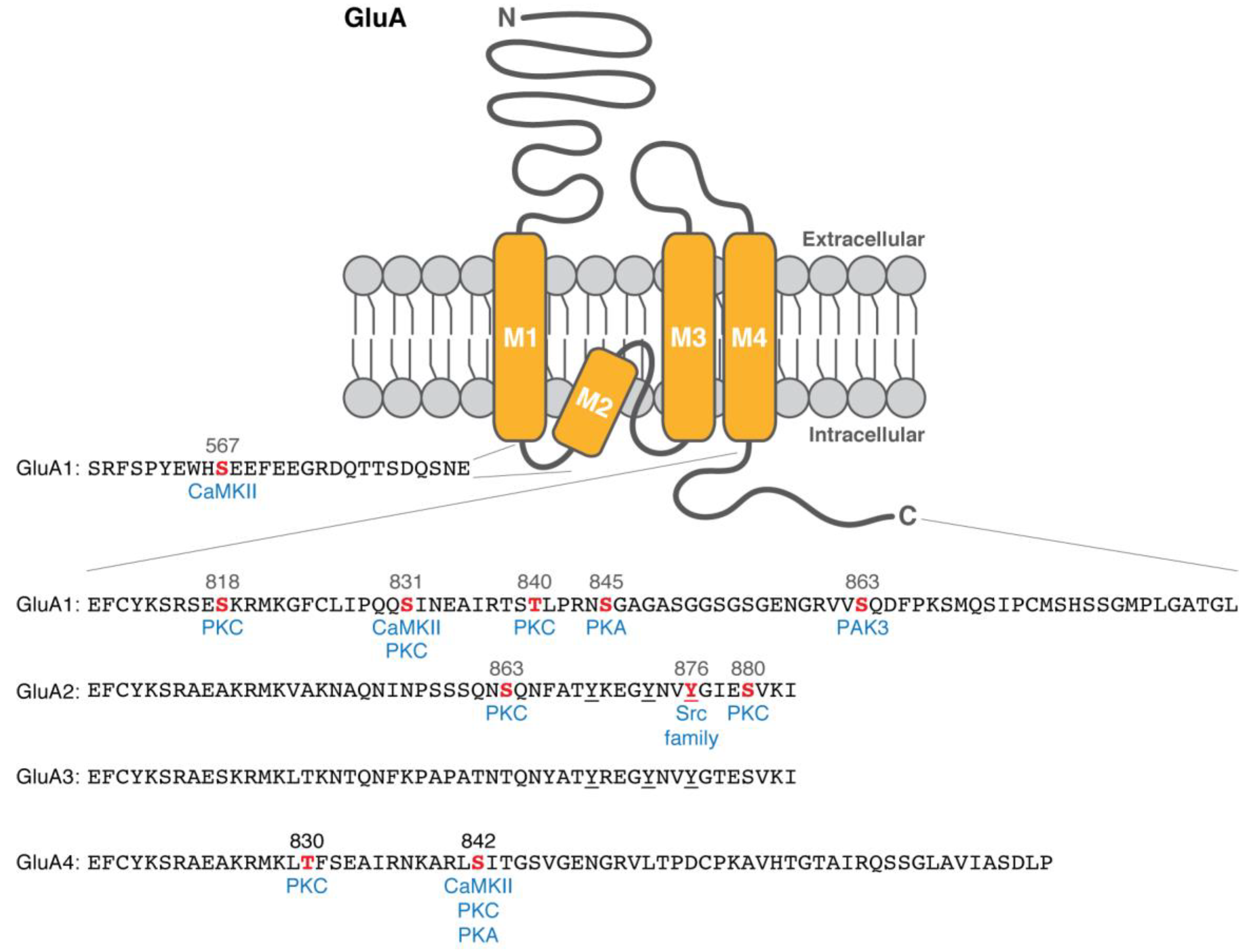


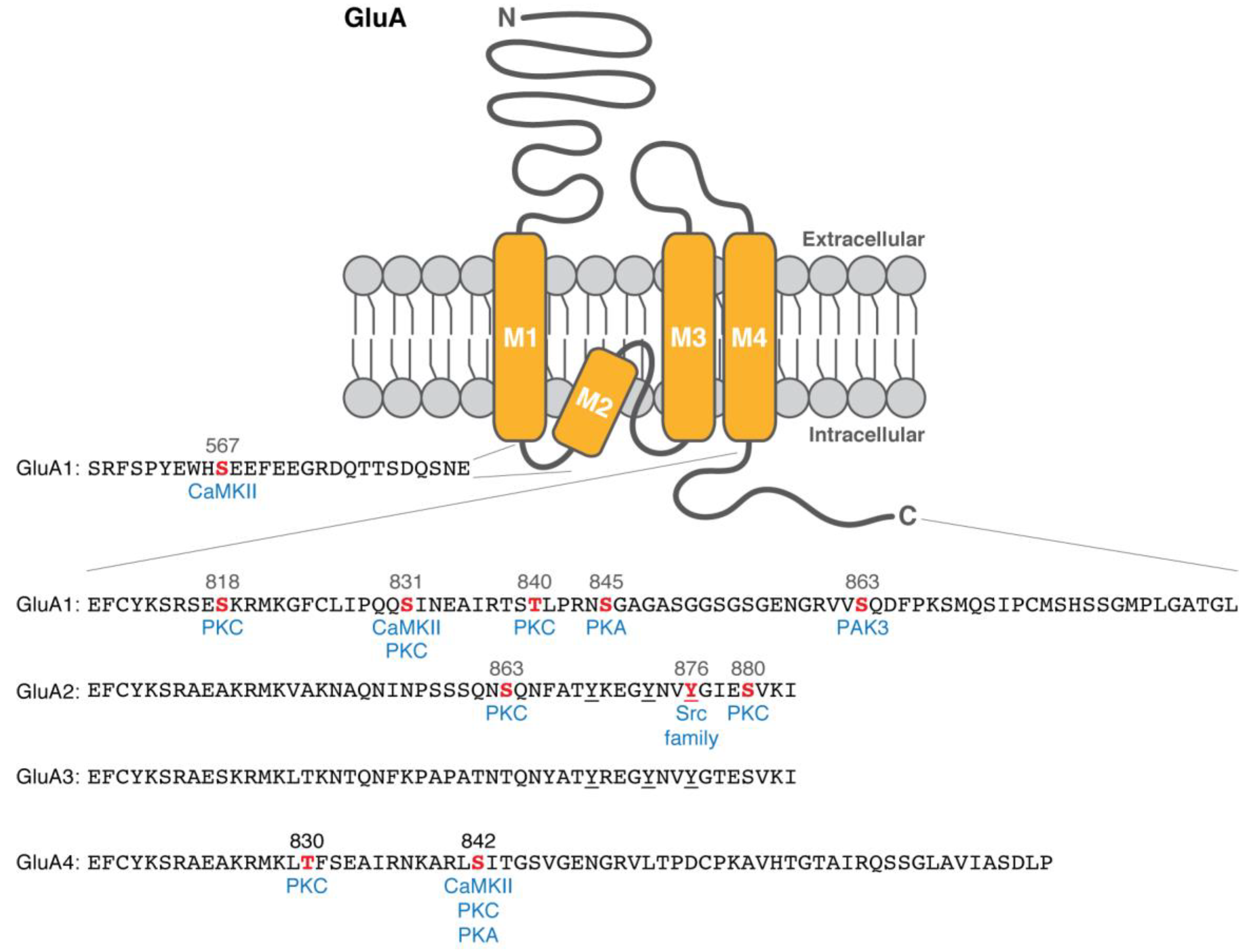{kind=link}
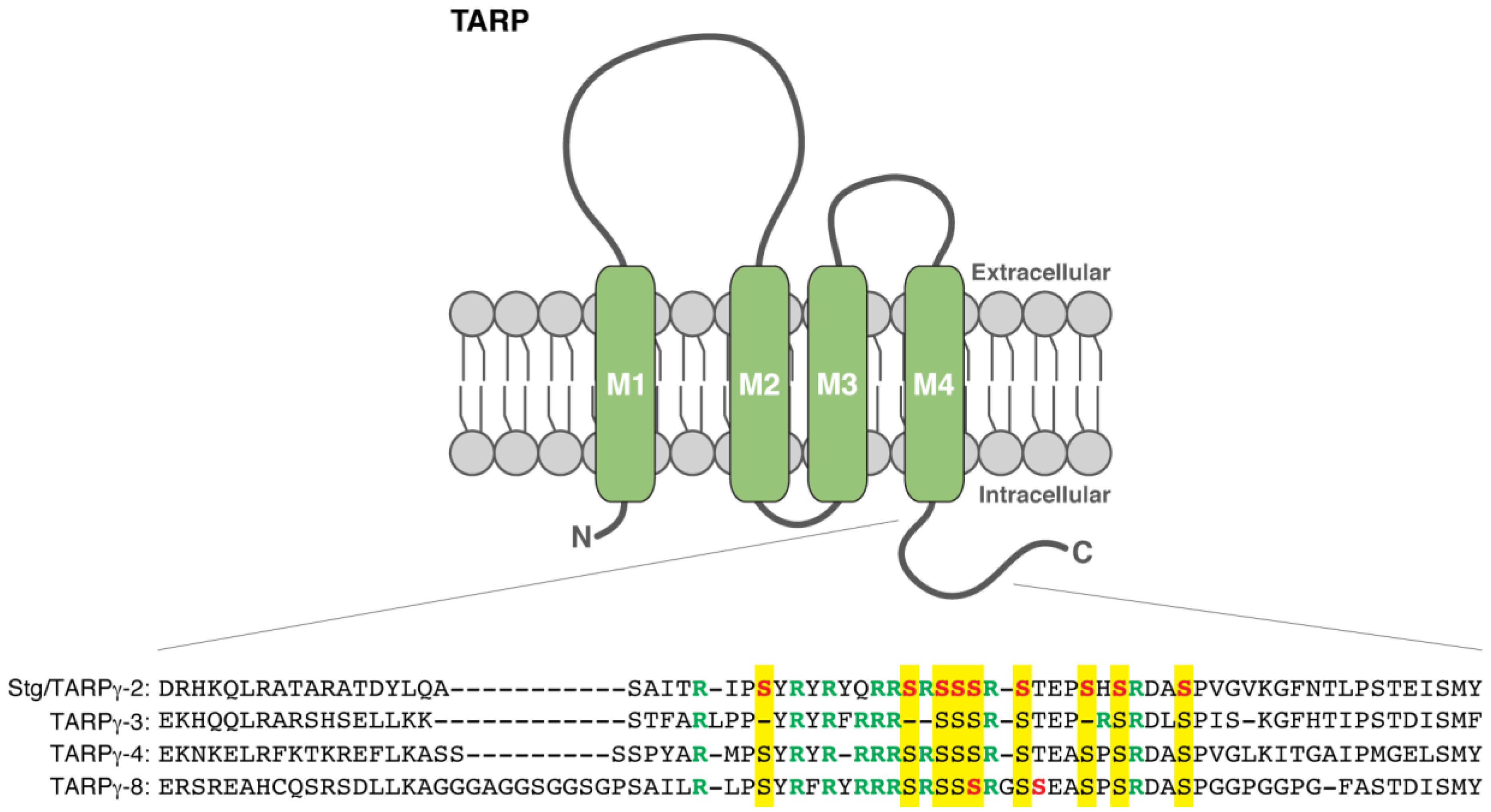{kind=link}
| Protein | Target Site | Kinase | Identification | Effect on the AMPAR Complex | Involvement in Synaptic Plasticity |
|---|---|---|---|---|---|
| GluA1 | Ser567 | CaMKII | In vitro; Phospho-specific antibody with rat hippocampal lysate [30] | Regulation of synaptic trafficking [30] | |
| Ser818 | PKC | In vitro; PKC activation of heterologous cells; Phospho-specific antibody with rat cortical lysate [25] | Enhancement of the weighted mean channel conductance [28] | A correlational increase upon chemical LTP and TBS [25] | |
| Ser831 | CaMKII | In vitro [17]; Co-expression of a constitutively active CaMKII in heterologous cells [19]; Phospho-specific antibody with rat hippocampal lysate [19,23] | Potentiation of AMPAR current [17]; Enhancement of channel conductance [20,21] | A correlational increase upon LTP [23] | |
| PKC | In vitro; PKC- and PKA-activation of heterologous cells [18] | ||||
| Thr840 | PKC | In vitro [26,27]; Phosphopeptide mapping of hippocampal slices [26]; Phospho-specific antibody with mouse hippocampal lysate [26,27] | Inhibition of PKA-induced AMPAR potentiation [27]; Enhancement of the weighted mean channel conductance [28] | A correlational change upon PKC activity [26,27] | |
| Ser845 | PKA | In vitro [18]; PKA activation of heterologous cells [18,19]; Phospho-specific antibody with rat hippocampal lysate [19,23] | Potentiation of AMPAR current [18]; Enhancement of channel opening probability [22] | A correlational decrease upon LTD [23] | |
| Ser863 | PAK3 | In vitro; Co-expression in heterologous cells; Phospho-specific antibody with cortical lysate [29] | Regulation of surface expression [29] | ||
| GluA2 | Ser863 | PKC | In vitro; PKC activation of heterologous cells; Phospho-specific antibody with cortical lysate [31] | ||
| Tyr876 | Lyn | Co-expression in heterologous cells [32] | Regulation of GRIP binding [32,33] | ||
| Tyr869, Tyr873, Tyr876 | Internalization of GluA2 subunits [32,34] | A correlational increase upon LTD [34] | |||
| Ser880 | PKC | In vitro; PKC activation of heterologous cells [31,33] | Regulation of GRIP binding [33] | Contribution to LTD in GluA2 KO cerebellar Purkinje cell cultures [13] | |
| GluA4 | Thr830 | PKC | In vitro [35] | ||
| Ser842 | CaMKII | In vitro [35] | Synaptic incorporation of GluA4 subunits [36] | A correlational increase upon PKA activation [36] | |
| PKC | In vitro [35] | ||||
| PKA | In vitro; PKC activation of heterologous cells [35] | ||||
| Stargazin/TARPγ-2 | Ser228, Ser237, Ser239, Ser240, Ser241, Ser243, Ser247, Ser249, Ser253 | CaMKII, PKC | Phosphopeptide mapping of heterologous cells and cortical neurons [37] | Dissociation of the cytoplasmic domain from the plasma membrane [38,39] | Contribution to LTP in hippocampal slice culture [37] |
| Thr321 | PKA, ERK2, p38 MAPK | In vitro [40] | Regulation of PSD-95 binding [40] | ||
| TARPγ-8 | Ser277, Ser281 | CaMKII | In vitro; Radio-Edman sequencing [41] | Enhancement of synaptic AMPAR activity [41] | A correlational increase upon chemical LTP [41] |
| CKAMP44/Shisa9 | N/A | PKC | In vitro; PKC activation of heterologous cells [42] |
| Protein | Phosphorylation Site (Mutated to Alanine Residues) | Effect on Synaptic Plasticity |
|---|---|---|
| GluA1 | Ser831, Thr838, Ser839, Thr840, and Ser845 | A reduction in LTP and LTD at adult stage [26] |
| Ser831 and Ser845 | A reduction in LTP and LTD at adult stage [43] | |
| Ser831 | Normal LTP and LTD [44] | |
| Ser845 | Abolished LTD [44] | |
| Stargazin/TARPγ-2 | Ser228, Ser237, Ser239, Ser240, Ser241, Ser243, Ser247, Ser249, and Ser253 | Normal LTP [41] |
| TARPγ-8 | Ser277 and Ser281 | A reduction in LTP [41] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J. Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity. Proteomes 2018, 6, 40. https://doi.org/10.3390/proteomes6040040
Park J. Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity. Proteomes. 2018; 6(4):40. https://doi.org/10.3390/proteomes6040040
Chicago/Turabian StylePark, Joongkyu. 2018. "Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity" Proteomes 6, no. 4: 40. https://doi.org/10.3390/proteomes6040040
APA StylePark, J. (2018). Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity. Proteomes, 6(4), 40. https://doi.org/10.3390/proteomes6040040