Fyn Regulates Binding Partners of Cyclic-AMP Dependent Protein Kinase A

 and
and 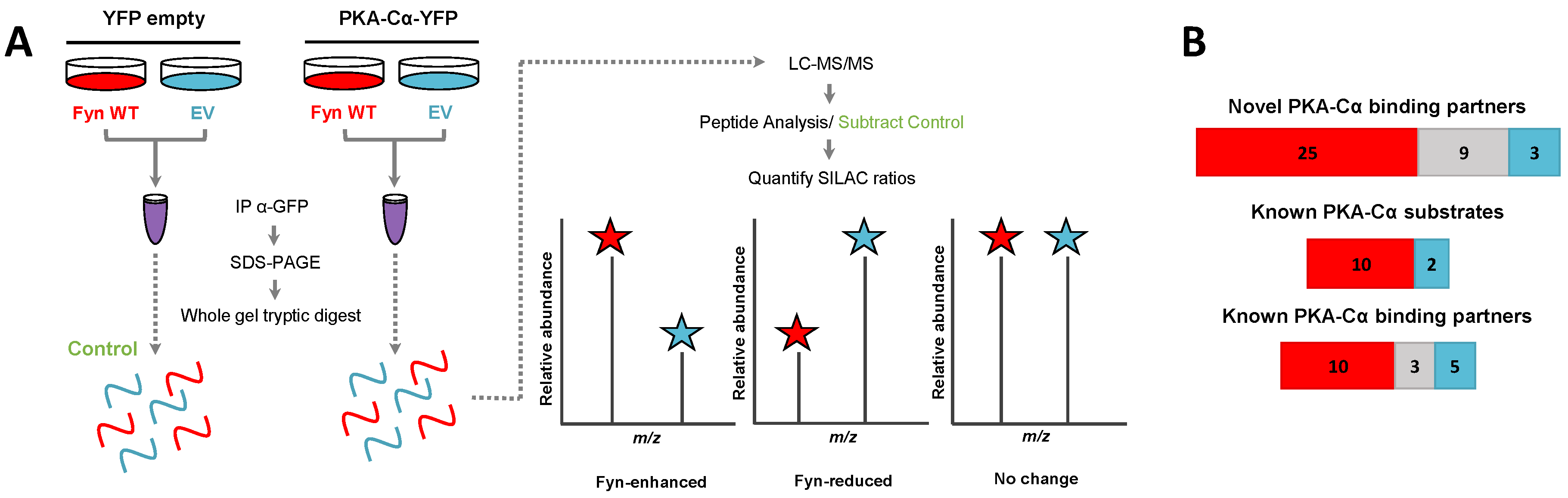{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmids and Antibodies
2.3. Cell Culture
2.4. Transfection and Immunoprecipitation for Mass Spectrometry
2.5. Peptide Processing for Mass Spectrometry
2.6. Transfection and Immunoprecipitation for Western Blotting
3. Results
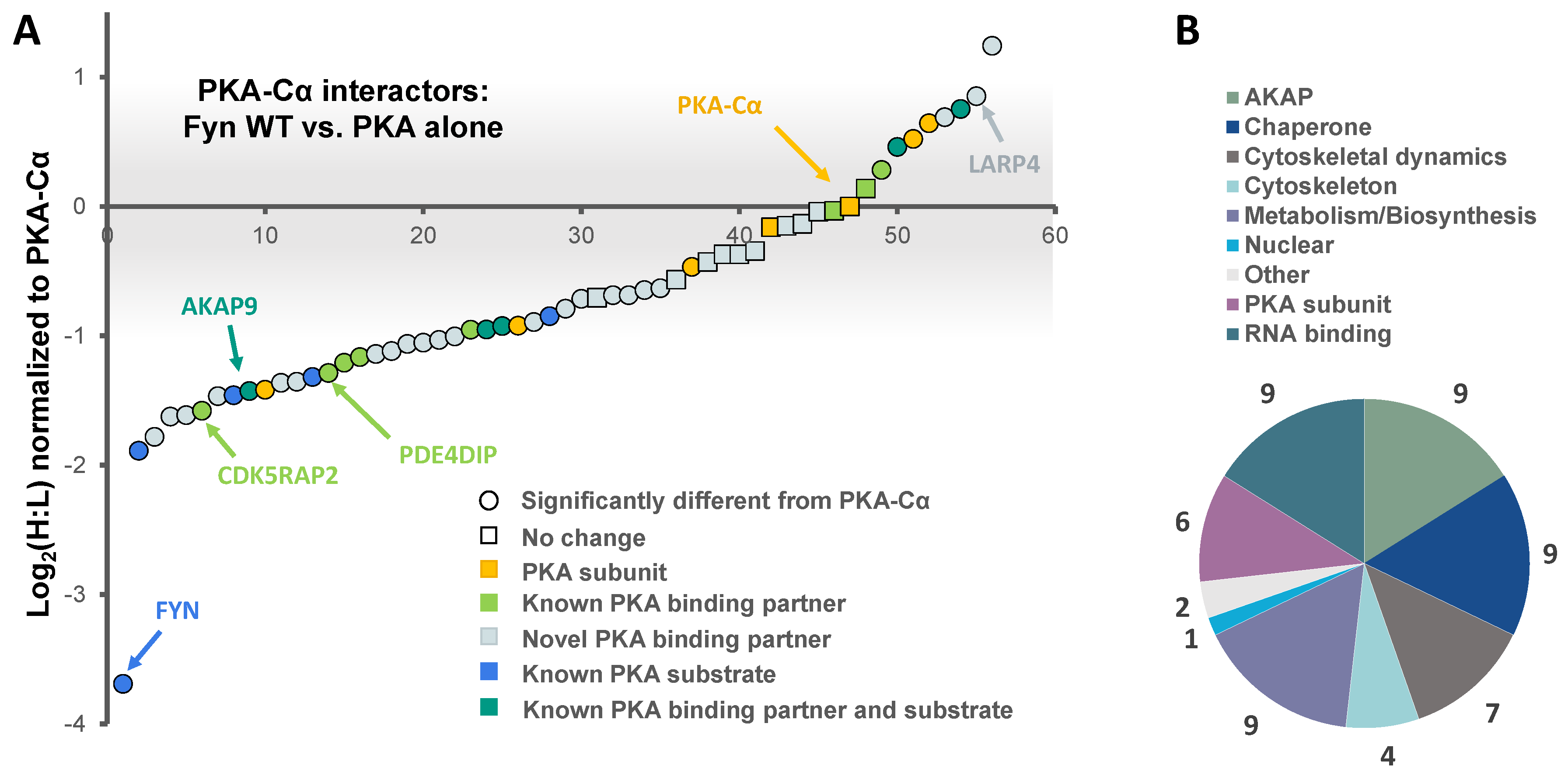
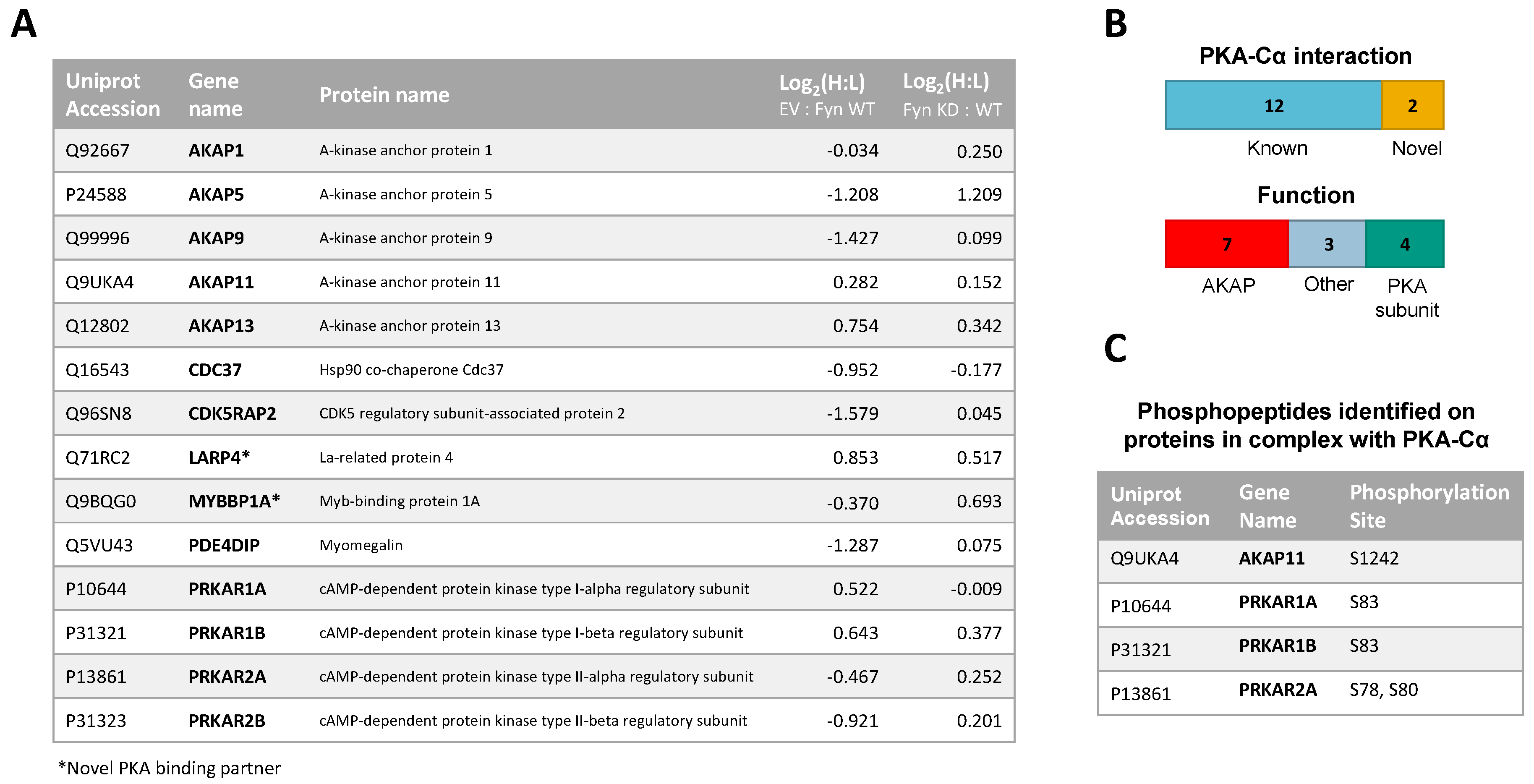
3.1. Identification of Fyn-Mediated PKA–Cα Binding Partners
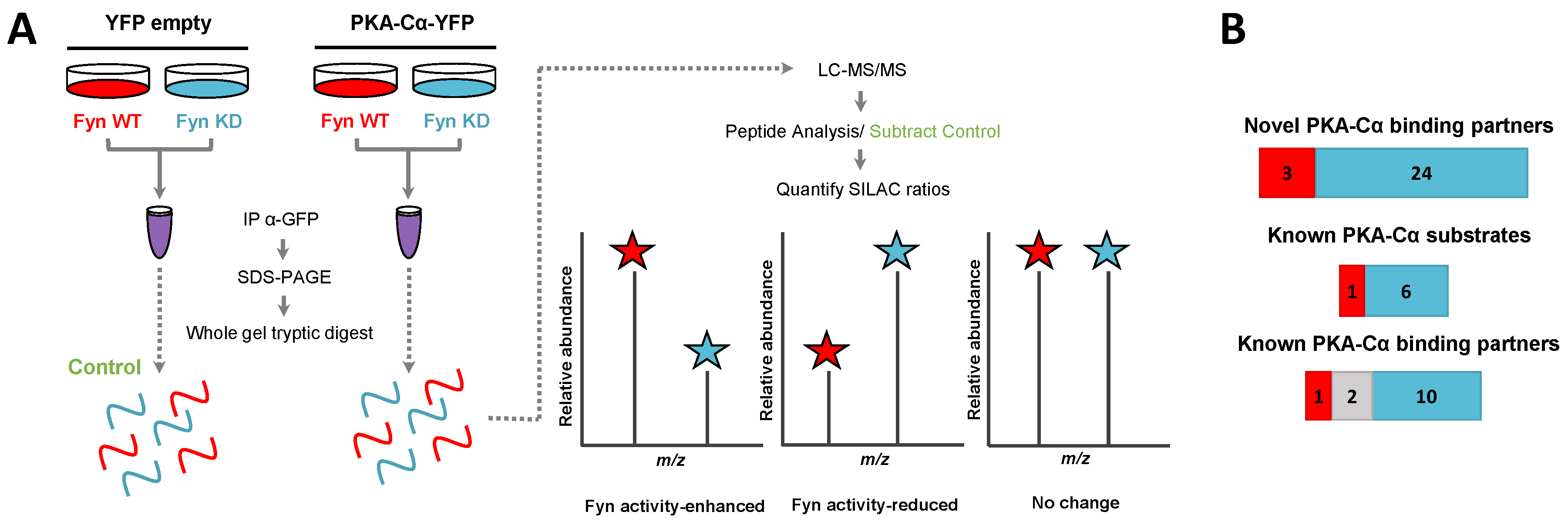
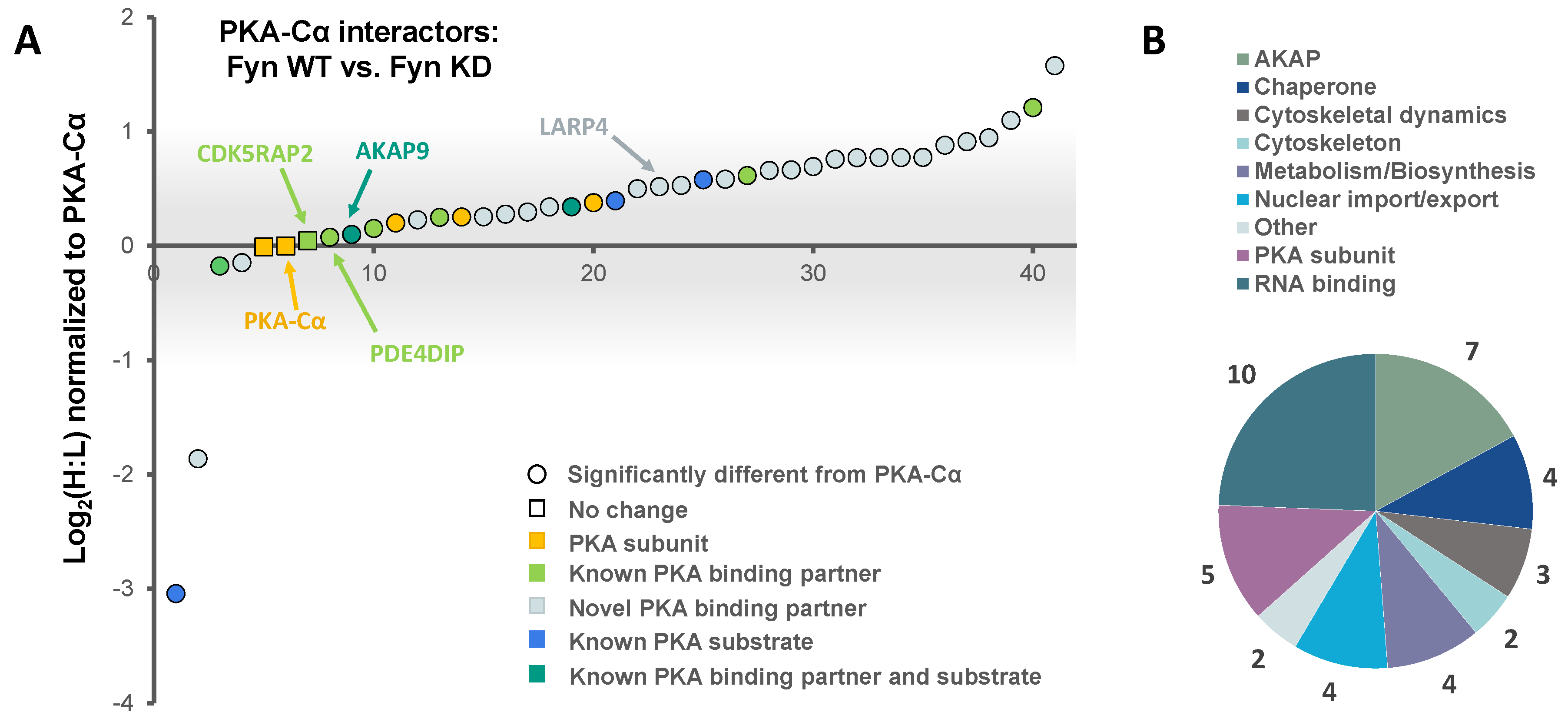
3.2. Effect of Fyn-Activity on Proteins in Complex with PKA–Cα
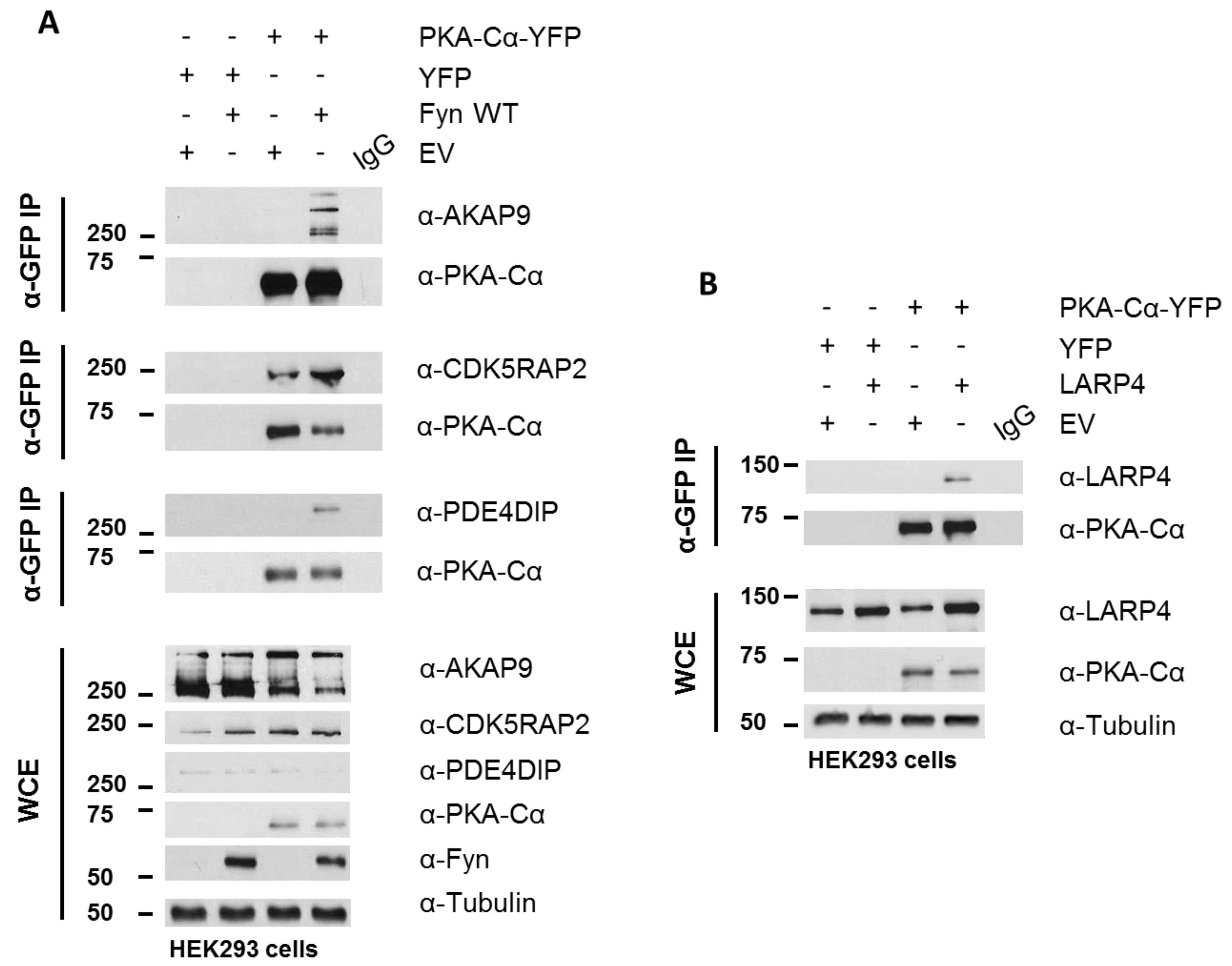
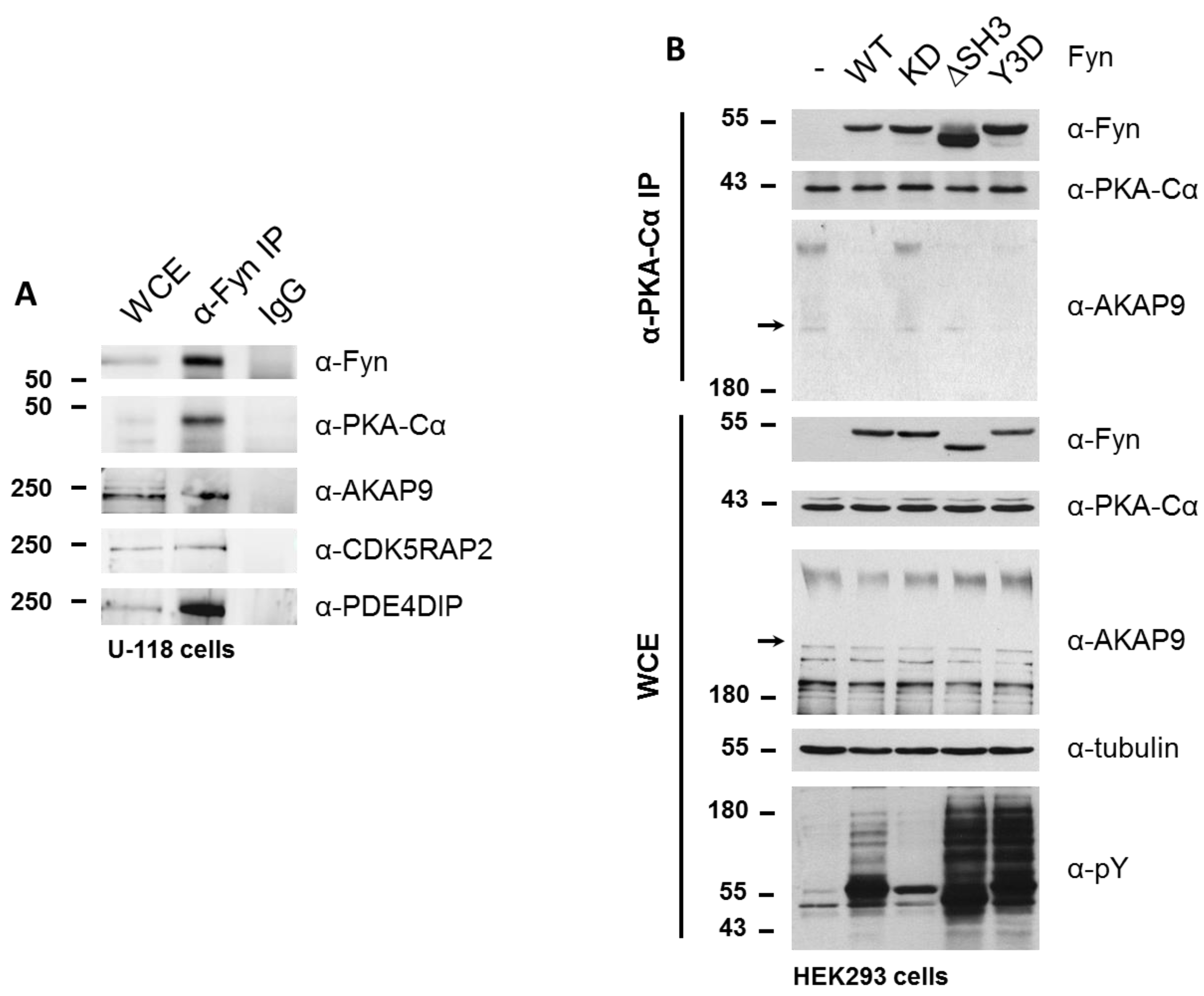
3.3. Biochemcial Validation of Fyn-Dependent Binding Partners
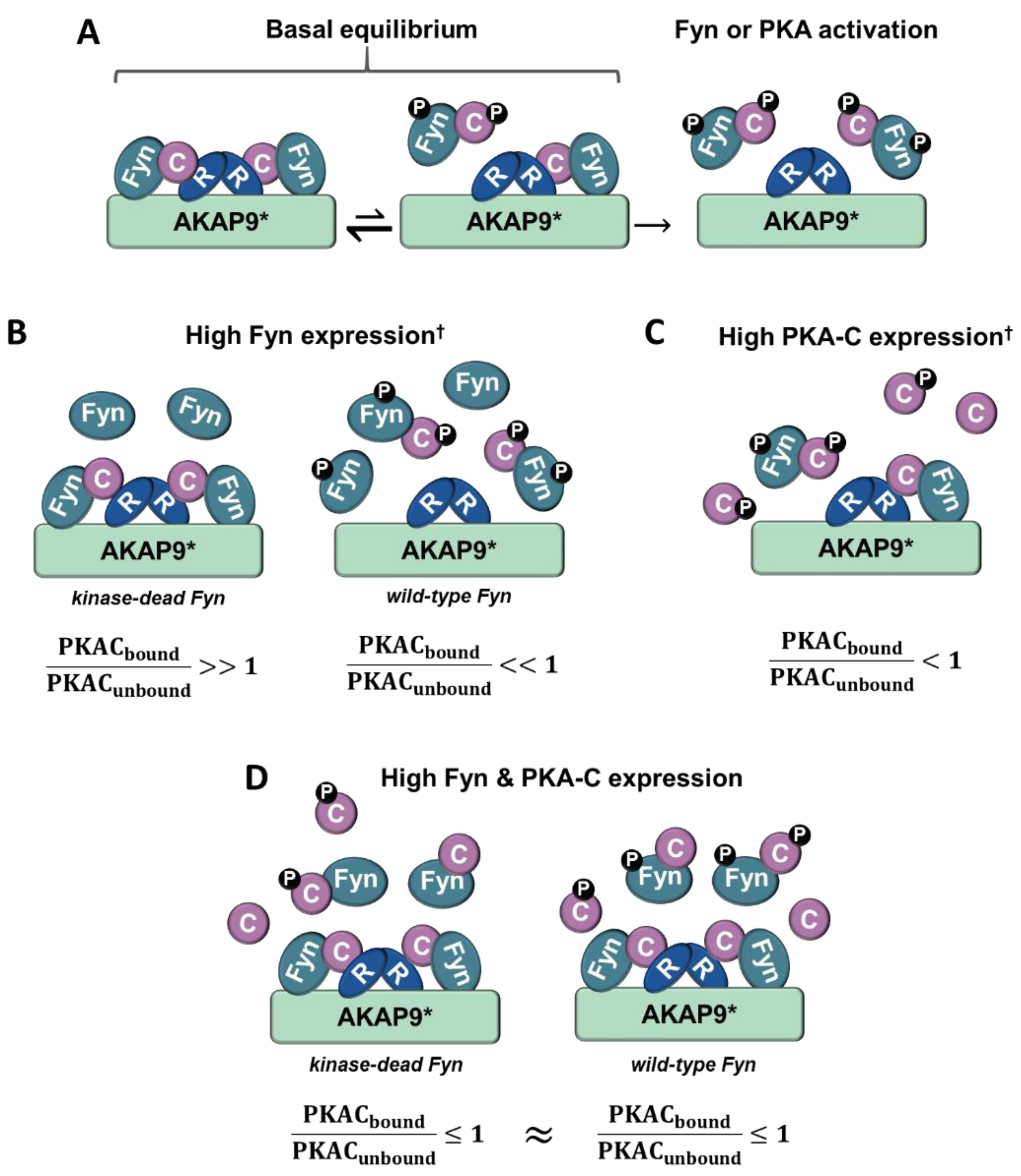
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kannan, N.; Haste, N.; Taylor, S.S.; Neuwald, A.F. The hallmark of agc kinase functional divergence is its c-terminal tail, a cis-acting regulatory module. Proc. Natl. Acad. Sci. USA 2007, 104, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Gerbaud, P.; Taskén, K.; Pidoux, G. Spatiotemporal regulation of cAMP signaling controls the human trophoblast fusion. Front. Pharmacol. 2015, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Jarnaess, E.; Taskén, K. Spatiotemporal Control of CAMP Signalling Processes by Anchored Signalling Complexes. Biochem. Soc. Trans. 2007, 35, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.T.; Chalasani, M.L.S.; Fazil, M.; Prasannan, P.; Kizhakeyil, A.; Wright, G.D.; Kelleher, D.; Verma, N.K. Centrosome-and golgi-localized protein kinase n-associated protein serves as a docking platform for protein kinase A signaling and microtubule nucleation in migrating t-cells. Front. Immunol. 2018, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, G.; Taskén, K. Specificity and spatial dynamics of protein kinase A signaling organized by a-kinase-anchoring proteins. J. Mol. Endocrinol. 2010, 44, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Raslan, Z.; Aburima, A.; Naseem, K.M. The spatiotemporal regulation of cAMP signaling in blood platelets—Old friends and new players. Front. Pharmacol. 2015, 6, 266. [Google Scholar] [CrossRef] [PubMed]
- Sample, V.; DiPilato, L.M.; Yang, J.H.; Ni, Q.; Saucerman, J.J.; Zhang, J. Regulation of nuclear pka revealed by spatiotemporal manipulation of cyclic amp. Nat. Chem. Biol. 2012, 8, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with rii-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar] [PubMed]
- Bendzunas, N.G.; Dörfler, S.; Autenrieth, K.; Bertinetti, D.; Machal, E.M.; Kennedy, E.J.; Herberg, F.W. Investigating PKA-RII specificity using analogs of the PKA: AKAP peptide inhibitor STAD-2. Bioorg. Med. Chem. 2018, 26, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Scott, J.D. Akap signaling complexes: Pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 2013, 34, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Herberg, F.W.; Maleszka, A.; Eide, T.; Vossebein, L.; Tasken, K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: Pka isoform specificity in AKAP binding1. J. Mol. Biol. 2000, 298, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.-S.; Wang, L.; Ma, Y.; Durick, K.; Perkins, G.; Deerinck, T.J.; Ellisman, M.H.; Taylor, S.S. NH2-terminal targeting motifs direct dual specificity A-kinase–anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J. Cell Biol. 1999, 145, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. Akap signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, H.; Vang, T.; Taskén, K. Protein kinase A intersects src signaling in membrane microdomains. J. Biol. Chem. 2003, 278, 17170–17177. [Google Scholar] [CrossRef] [PubMed]
- Krapf, D.; Arcelay, E.; Wertheimer, E.V.; Sanjay, A.; Pilder, S.H.; Salicioni, A.M.; Visconti, P.E. Inhibition of ser/thr phosphatases induces capacitation-associated signaling in the presence of src kinase inhibitors. J. Biol. Chem. 2010, 285, 7977–7985. [Google Scholar] [CrossRef] [PubMed]
- Beristain, A.; Molyneux, S.; Joshi, P.; Pomroy, N.; Di Grappa, M.; Chang, M.; Kirschner, L.S.; Privé, G.; Pujana, M.; Khokha, R. PKA signaling drives mammary tumorigenesis through Src. Oncogene 2015, 34, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.M.; Stork, P.J. PKA phosphorylation of src mediates cAMP’s inhibition of cell growth via Rap1. Mol. Cell 2002, 9, 85–94. [Google Scholar] [CrossRef]
- Caldwell, G.B.; Howe, A.K.; Nickl, C.K.; Dostmann, W.R.; Ballif, B.A.; Deming, P.B. Direct modulation of the protein kinase A catalytic subunit α by growth factor receptor tyrosine kinases. J. Cell. Biochem. 2012, 113, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.G.; Oh, H.J.; Cho, H.-S.; Chun, J.S.; Marcantonio, E.E.; Song, W.K. Phosphorylation of ser 21 in fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J. Cell. Physiol. 2011, 226, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed]
- Deming, P.B.; Ballif, B.A. unpublished observations. 2018.
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gaidamakov, S.A.; Xie, J.; Lee, J.; Martino, L.; Kozlov, G.; Crawford, A.K.; Russo, A.N.; Conte, M.R.; Gehring, K.; et al. La-related protein 4 binds poly(A), interacts with the poly(A)-binding protein mlle domain via a variant PAM2w motif, and can promote mrna stability. Mol. Cell. Biol. 2011, 31, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.E.; Mann, J.E.; Corwin, T.; Fulton, Z.W.; Hao, J.M.; Maniscalco, J.F.; Kenney, M.C.; Roque, K.M.R.; Chapdelaine, E.F.; Stelzl, U. Novel autophosphorylation sites of src family kinases regulate kinase activity and SH2 domain-binding capacity. FEBS Lett. 2016, 590, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Schmoker, A.M.; Weinert, J.L.; Kellett, K.J.; Johnson, H.E.; Joy, R.M.; Weir, M.E.; Ebert, A.M.; Ballif, B.A. Dynamic multi-site phosphorylation by Fyn and Abl drives the interaction between CRKl and the novel scaffolding receptors DCBLD1 and DCBLD2. Biochem. J. 2017, 474, 3963–3984. [Google Scholar] [CrossRef] [PubMed]
- Bakalarski, C.E.; Elias, J.E.; Villén, J.; Haas, W.; Gerber, S.A.; Everley, P.A.; Gygi, S.P. The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J. Proteome Res. 2008, 7, 4756–4765. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. Phosphositeplus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Rios, R.M. The centrosome–golgi apparatus nexus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, S.; Flemyng, E.; Shen, J.; Conte, M.R.; Ridley, A.J. The rna-binding protein larp4 regulates cancer cell migration and invasion. Cytoskeleton 2016, 73, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Abuin, L.; Cotecchia, S.; Pansier, L. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004, 23, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Ozer, R.S.; Halpain, S. Phosphorylation-dependent localization of microtubule-associated protein map2c to the actin cytoskeleton. Mol. Biol. Cell 2000, 11, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Melková, K.; Zapletal, V.; Jansen, S.; Nomilner, E.; Zachrdla, M.; Hritz, J.; Nováček, J.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Functionally specific binding regions of microtubule-associated protein 2c exhibit distinct conformations and dynamics. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zamora-Leon, S.P.; Lee, G.; Davies, P.; Shafit-Zagardo, B. Binding of Fyn to MAP-2c through an SH3 binding domain regulation of the interaction by ERK2. J. Biol. Chem. 2001, 276, 39950–39958. [Google Scholar] [CrossRef] [PubMed]
- Uys, G.M.; Ramburan, A.; Loos, B.; Kinnear, C.J.; Korkie, L.J.; Mouton, J.; Riedemann, J.; Moolman-Smook, J.C. Myomegalin is a novel A-kinase anchoring protein involved in the phosphorylation of cardiac myosin binding protein C. BMC Cell Biol. 2011, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Roubin, R.; Acquaviva, C.; Chevrier, V.; Sedjaï, F.; Zyss, D.; Birnbaum, D.; Rosnet, O. Myomegalin is necessary for the formation of centrosomal and golgi-derived microtubules. Biol. Open 2013, 2, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Pahlke, G.; Salanova, M.; Zhang, G.; Wang, S.; Coletti, D.; Onuffer, J.; Jin, S.-L.C.; Conti, M. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 2001, 276, 11189–11198. [Google Scholar] [CrossRef] [PubMed]
- Terrin, A.; Monterisi, S.; Stangherlin, A.; Zoccarato, A.; Koschinski, A.; Surdo, N.C.; Mongillo, M.; Sawa, A.; Jordanides, N.E.; Mountford, J.C.; et al. PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J. Cell Biol. 2012, 198, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Cep proteins: The knights of centrosome dynasty. Protoplasma 2013, 250, 965–983. [Google Scholar] [CrossRef] [PubMed]
- Colello, D.; Mathew, S.; Ward, R.; Pumiglia, K.; LaFlamme, S.E. Integrins regulate the microtubule nucleating activity of the centrosome through MEK/ERK signaling. J. Biol. Chem. 2012, 287, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Walker-Gray, R.; Stengel, F.; Gold, M.G. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. USA 2017, 114, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Guarguaglini, G.; Duncan, P.I.; Stierhof, Y.D.; Holmström, T.; Duensing, S.; Nigg, E.A. The forkhead-associated domain protein Cep170 interacts with polo-like kinase 1 and serves as a marker for mature centrioles. Mol. Biol. Cell 2005, 16, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Rogers, G.C.; Sharp, D.J.; Vale, R.D. Drosophila EB1 is important for proper assembly, dynamics, and positioning of the mitotic spindle. J. Cell Biol. 2002, 158, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Bouguenina, H.; Salaun, D.; Mangon, A.; Muller, L.; Baudelet, E.; Camoin, L.; Tachibana, T.; Cianférani, S.; Audebert, S.; Verdier-Pinard, P. Eb1-binding–myomegalin protein complex promotes centrosomal microtubules functions. Proc. Natl. Acad. Sci. USA 2017, 114, E10687–E10696. [Google Scholar] [CrossRef] [PubMed]
- Salpingidou, G.; Smertenko, A.; Hausmanowa-Petrucewicz, I.; Hussey, P.J.; Hutchison, C.J. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 2007, 178, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Trevor, K.T.; McGuire, J.G.; Leonova, E.V. Association of vimentin intermediate filaments with the centrosome. J. Cell Sci. 1995, 108, 343–356. [Google Scholar] [PubMed]
- Varticovski, L.; Chahwala, S.B.; Whitman, M.; Cantley, L.; Schindler, D.; Chow, E.P.; Sinclair, L.K.; Pepinsky, R.B. Location of sites in human lipocortin i that are phosphorylated by protein tyrosine kinases and protein kinases A and C. Biochemistry 1988, 27, 3682–3690. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.S.; Traganos, F.; Darzynkiewicz, Z.; Lee, M.Y. Phosphorylation of Riα by Cyclin-Dependent kinase CDK 2/Cyclin E modulates the dissociation of the RIα-RFC40 complex. Cell Cycle 2006, 5, 654–661. [Google Scholar] [CrossRef]
- Haushalter, K.J.; Casteel, D.E.; Raffeiner, A.; Stefan, E.; Patel, H.H.; Taylor, S.S. Phosphorylation of protein kinase A (PKA) regulatory subunit riα by protein kinase G (PKG) primes pka for catalytic activity in cells. J. Biol. Chem. 2018, 293, 4411–4421. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmoker, A.M.; Barritt, S.A.; Weir, M.E.; Mann, J.E.; Hogan, T.C.; Ballif, B.A.; Deming, P.B. Fyn Regulates Binding Partners of Cyclic-AMP Dependent Protein Kinase A. Proteomes 2018, 6, 37. https://doi.org/10.3390/proteomes6040037
Schmoker AM, Barritt SA, Weir ME, Mann JE, Hogan TC, Ballif BA, Deming PB. Fyn Regulates Binding Partners of Cyclic-AMP Dependent Protein Kinase A. Proteomes. 2018; 6(4):37. https://doi.org/10.3390/proteomes6040037
Chicago/Turabian StyleSchmoker, Anna M., Samuel A. Barritt, Marion E. Weir, Jacqueline E. Mann, Tyler C. Hogan, Bryan A. Ballif, and Paula B. Deming. 2018. "Fyn Regulates Binding Partners of Cyclic-AMP Dependent Protein Kinase A" Proteomes 6, no. 4: 37. https://doi.org/10.3390/proteomes6040037
APA StyleSchmoker, A. M., Barritt, S. A., Weir, M. E., Mann, J. E., Hogan, T. C., Ballif, B. A., & Deming, P. B. (2018). Fyn Regulates Binding Partners of Cyclic-AMP Dependent Protein Kinase A. Proteomes, 6(4), 37. https://doi.org/10.3390/proteomes6040037