Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias


Abstract
1. Introduction
2. LC-MS/MS Strategies
2.1. Data-Dependent LC-MS/MS
2.2. Targeted LC-MS/MS Acquisition
2.3. Data-Independent Acquisition
2.4. Candidate Disease Markers from LC-MS/MS Studies
3. Capture-Based Strategies
3.1. Multiplexed Immunoassays
3.2. Adaptations of Standard Capture Methods
3.3. Ultrasensitive Detection Methods
3.4. Candidate Disease Markes from Capture-Based Studies
4. Considerations for Accurate and Reproducible Findings
4.1. Preanalytical Effects
4.2. Matrix Effects
4.3. Data Processing
4.4. Multisite Variability
5. Future Directions
Funding
Conflicts of Interest
References
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Sheikh-Bahaei, N.; Sajjadi, S.A.; Manavaki, R.; Gillard, J.H. Imaging Biomarkers in Alzheimer’s Disease: A Practical Guide for Clinicians. J. Alzheimer’s Dis. Rep. 2017, 1, 71–88. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H.; Fagan, A.M. Fluid biomarkers in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006221. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Robert Snodgrass, S.; Johanson, C.E. A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp. Neurol. 2015, 273, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Applying fluid biomarkers to Alzheimer’s disease. Am. J. Physiol.-Cell Physiol. 2017, 313, C3–C10. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.M.; Carrillo, M.C.; Grodstein, F.; Henriksen, K.; Jeromin, A.; Lovestone, S.; Mielke, M.M.; O’Bryant, S.; Sarasa, M.; Sjøgren, M.; et al. Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; van der Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s Dement. 2017, 8, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Duits, F.H.; Martinez-Lage, P.; Paquet, C.; Engelborghs, S.; Lleó, A.; Hausner, L.; Molinuevo, J.L.; Stomrud, E.; Farotti, L.; Ramakers, I.H.; et al. Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study. Alzheimer’s Dement. 2016, 12, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Baird, A.L.; Westwood, S.; Hye, A.; Dobson, R.; Thambisetty, M.; Lovestone, S. A Decade of Blood Biomarkers for Alzheimer’s Disease Research: An Evolving Field, Improving Study Designs, and the Challenge of Replication. J. Alzheimer’s Dis. 2018, 62, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; McTernan, P.G.; Schraw, T.; Kos, K.; O’Hare, J.P.; Ahima, R.; Kumar, S.; Scherer, P.E. Adiponectin complexes in human cerebrospinal fluid: Distinct complex distribution from serum. Diabetologia 2007, 50, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. ADNI Investigators Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.H.; Rissin, D.M.; Kan, C.W.; Fournier, D.R.; Piech, T.; Campbell, T.G.; Meyer, R.E.; Fishburn, M.W.; Cabrera, C.; Patel, P.P.; et al. The Simoa HD-1 Analyzer. J. Lab. Autom. 2016, 21, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin. Chem. Lab. Med. 2016, 54, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Candia, J.; Cheung, F.; Kotliarov, Y.; Fantoni, G.; Sellers, B.; Griesman, T.; Huang, J.; Stuccio, S.; Zingone, A.; Ryan, B.M.; et al. Assessment of Variability in the SOMAscan Assay. Sci. Rep. 2017, 7, 14248. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, G.F.; Shah, H.P.; Kenten, J.H.; Leland, J.; Kamin, R.A.; Link, J.; Peterman, J.; Powell, M.J.; Shah, A.; Talley, D.B. Electrochemiluminescence detection for development of immunoassays and DNA probe assays for clinical diagnostics. Clin. Chem. 1991, 37, 1534–1539. [Google Scholar] [PubMed]
- Gross, E.M.; Maddipati, S.S.; Snyder, S.M. A review of electrogenerated chemiluminescent biosensors for assays in biological matrices. Bioanalysis 2016, 8, 2071–2089. [Google Scholar] [CrossRef] [PubMed]
- Ledger, K.S.; Agee, S.J.; Kasaian, M.T.; Forlow, S.B.; Durn, B.L.; Minyard, J.; Lu, Q.A.; Todd, J.; Vesterqvist, O.; Burczynski, M.E. Analytical validation of a highly sensitive microparticle-based immunoassay for the quantitation of IL-13 in human serum using the Erenna® immunoassay system. J. Immunol. Methods 2009, 350, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, B.A.; Carlyle, B.C.; Koenig, A.M.; Shaw, L.M.; Trojanowski, J.Q.; Wolk, D.A.; Locascio, J.J.; Arnold, S.E. The technical reliability and biotemporal stability of cerebrospinal fluid biomarkers for profiling multiple pathophysiologies in Alzheimer’s disease. PLoS ONE 2018, 13, e0193707. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhu, W.; Cui, F.; Yang, F.; Chen, Z.; Ling, L.; Huang, X. Measurement of cystatin C levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Int. J. Clin. Exp. Pathol. 2015, 8, 5419–5426. [Google Scholar] [PubMed]
- Blennow, K.; Davidsson, P.; Wallin, A.; Ekman, R. Chromogranin A in cerebrospinal fluid: A biochemical marker for synaptic degeneration in Alzheimer’s disease? Dementia 1995, 6, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Yang, J.; Chambers, A.G.; Simon, R.; Hardie, D.B.; Borchers, C.H. Multiplexed MRM with Internal Standards for Cerebrospinal Fluid Candidate Protein Biomarker Quantitation. J. Proteome Res. 2014, 13, 3733–3747. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, T.; Qian, W.-J.; Petyuk, V.A.; Smith, R.D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid Chromatography Mass Spectrometry-Based Proteomics: Biological and Technological Aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef] [PubMed]
- Drabik, A. Quantitative Measurements in Proteomics: Mass Spectrometry. Proteom. Profiling Anal. Chem. 2016, 145–160. [Google Scholar] [CrossRef]
- Mostovenko, E.; Hassan, C.; Rattke, J.; Deelder, A.M.; van Veelen, P.A.; Palmblad, M. Comparison of peptide and protein fractionation methods in proteomics. EuPA Open Proteom. 2013, 1, 30–37. [Google Scholar] [CrossRef]
- Bauer, M.; Ahrné, E.; Baron, A.P.; Glatter, T.; Fava, L.L.; Santamaria, A.; Nigg, E.A.; Schmidt, A. Assessment of current mass spectrometric workflows for the quantification of low abundant proteins and phosphorylation sites. Data Br. 2015, 5, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bubis, J.A.; Levitsky, L.I.; Ivanov, M.V.; Tarasova, I.A.; Gorshkov, M.V. Comparative evaluation of label-free quantification methods for shotgun proteomics. Rapid Commun. Mass Spectrom. 2017, 31, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996, 379, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Clauser, K.R.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol. Cell. Proteom. 2010, 9, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Vega-Montoto, L.; Rudnick, P.A.; Variyath, A.M.; Ham, A.-J.L.; Bunk, D.M.; Kilpatrick, L.E.; Billheimer, D.D.; Blackman, R.K.; Cardasis, H.L.; et al. Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography-Tandem Mass Spectrometry. J. Proteome Res. 2010, 9, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.; Branca, R.M.M.; Lehtiö, J.; Forshed, J. Quantitative accuracy in mass spectrometry based proteomics of complex samples: The impact of labeling and precursor interference. J. Proteom. 2014, 96, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC−MS/MS. J. Proteome Res. 2011, 10, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, B.C.; Kitchen, R.R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Sestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, J.A.; Noirel, J.; Brown, J.E.; Wright, P.C.; Evans, C.A. Quantitation with chemical tagging reagents in biomarker studies. PROTEOMICS—Clin. Appl. 2015, 9, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lapek, J.D.; Greninger, P.; Morris, R.; Amzallag, A.; Pruteanu-Malinici, I.; Benes, C.H.; Haas, W. Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. 2017, 35, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Latosinska, A.; Vougas, K.; Makridakis, M.; Klein, J.; Mullen, W.; Abbas, M.; Stravodimos, K.; Katafigiotis, I.; Merseburger, A.S.; Zoidakis, J.; et al. Comparative Analysis of Label-Free and 8-Plex iTRAQ Approach for Quantitative Tissue Proteomic Analysis. PLoS ONE 2015, 10, e0137048. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Adams, R.M.; Chourey, K.; Hurst, G.B.; Hettich, R.L.; Pan, C. Systematic Comparison of Label-Free, Metabolic Labeling, and Isobaric Chemical Labeling for Quantitative Proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Alvarez, S.; Hicks, L.M. Comprehensive Comparison of iTRAQ and Label-free LC-Based Quantitative Proteomics Approaches Using Two Chlamydomonas reinhardtii Strains of Interest for Biofuels Engineering. J. Proteome Res. 2012, 11, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; Gygi, S.P. A comprehensive proteomic and phosphoproteomic analysis of yeast deletion mutants of 14-3-3 orthologs and associated effects of rapamycin. Proteomics 2015, 15, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.L.; Heslegrave, A.; Mitra, V.; Zetterberg, H.; Pocock, J.M.; Ward, M.A.; Pike, I. Combined tissue and fluid proteomics with Tandem Mass Tags to identify low-abundance protein biomarkers of disease in peripheral body fluid: An Alzheimer’s Disease case study. Rapid Commun. Mass Spectrom. 2017, 31, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bellei, E.; Bergamini, S.; Monari, E.; Fantoni, L.I.; Cuoghi, A.; Ozben, T.; Tomasi, A. High-abundance proteins depletion for serum proteomic analysis: Concomitant removal of non-targeted proteins. Amino Acids 2011, 40, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Günther, R.; Krause, E.; Schümann, M.; Blasig, I.E.; Haseloff, R.F. Depletion of highly abundant proteins from human cerebrospinal fluid: A cautionary note. Mol. Neurodegener. 2015, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Gillette, M.A.; Mertins, P.; Clauser, K.R.; Mani, D.R.; Kuhn, E.W.; Farrell, L.A.; Gerszten, R.E.; Carr, S.A. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteom. 2015, 14, 2375–2393. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wu, J.; Karl, J.; Liao, H.; Zolg, W.; Guild, B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 2004, 4, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full Dynamic Range Proteome Analysis of S. cerevisiae by Targeted Proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Gillette, M.A.; Carr, S.A. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 2013, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Desiderio, D.M. Peptide quantification by tandem mass spectrometry. Mass Spectrom. Rev. 1996, 15, 213–240. [Google Scholar] [CrossRef]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Liebler, D.C.; Zimmerman, L.J. Targeted quantitation of proteins by mass spectrometry. Biochemistry 2013, 52, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Frederick, K. SWATH-MS: Data Acquisition and Analysis. Proteom. Profiling Anal. Chem. 2016, 161–173. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111–016717. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.-J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Bland, P.; Kogata, N.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. SWATH mass spectrometry as a tool for quantitative profiling of the matrisome. J. Proteome 2018. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.-C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.-C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.S.; Egertson, J.D.; Bollinger, J.G.; Searle, B.C.; Payne, S.H.; Noble, W.S.; MacCoss, M.J. PECAN: Library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods 2017, 14, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; Villen, J.; MacCoss, M.J. Comprehensive peptide quantification for data independent acquisition mass spectrometry using chromatogram libraries. bioRxiv 2018, 277822. [Google Scholar] [CrossRef]
- Schilling, B.; Gibson, B.W.; Hunter, C.L. Generation of High-Quality SWATH® Acquisition Data for Label-free Quantitative Proteomics Studies Using TripleTOF® Mass Spectrometers. Methods Mol. Biol. 2017, 1550, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC–SWATH–MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Muenchhoff, J.; Poljak, A.; Song, F.; Raftery, M.; Brodaty, H.; Duncan, M.; McEvoy, M.; Attia, J.; Schofield, P.W.; Sachdev, P.S. Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease across two independent cohorts. J. Alzheimer’s Dis. 2015, 43, 1355–1373. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Kiddle, S.J.; Graf, J.; Ward, M.; Baird, A.L.; Hye, A.; Westwood, S.; Wong, K.V.; Dobson, R.J.; Rabinovici, G.D.; et al. Blood protein predictors of brain amyloid for enrichment in clinical trials? Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2015, 1, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Westwood, S.; Leoni, E.; Hye, A.; Lynham, S.; Khondoker, M.R.; Ashton, N.J.; Kiddle, S.J.; Baird, A.L.; Sainz-Fuertes, R.; Leung, R.; et al. Blood-Based Biomarker Candidates of Cerebral Amyloid Using PiB PET in Non-Demented Elderly. J. Alzheimer’s Dis. 2016, 52, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Poljak, A.; Kochan, N.A.; Raftery, M.; Brodaty, H.; Smythe, G.A.; Sachdev, P.S. Plasma protein profiling of Mild Cognitive Impairment and Alzheimer’s disease using iTRAQ quantitative proteomics. Proteome Sci. 2014, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.; Schrag, M.; Mueller, C.; Zhou, W.; Crofton, A.; Petersen, F.; Dickson, A.; Kirsch, W.M. Assessing Candidate Serum Biomarkers for Alzheimer’s Disease: A Longitudinal Study. J. Alzheimer’s Dis. 2012, 30, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.; Grant, M.; Creese, A.J.; Mangialasche, F.; Cecchetti, R.; Cooper, H.J.; Mecocci, P.; Aldred, S. Plasma Levels of Complement 4a Protein are Increased in Alzheimer’s Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lee, A.; Nouwens, A.; Henderson, R.D.; McCombe, P.A. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph. Lateral Scler. Frontotemporal Degener. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, I.; Noguchi, M.; Arito, M.; Sato, T.; Omoteyama, K.; Maedomari, M.; Hasegawa, H.; Suematsu, N.; Okamoto, K.; Kato, T.; et al. Serum peptides as candidate biomarkers for dementia with Lewy bodies. Int. J. Geriatr. Psychiatry 2015, 30, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Wojcik, J.; Núñez Galindo, A.; Corthésy, J.; Cominetti, O.; Oikonomidi, A.; Henry, H.; Migliavacca, E.; Bowman, G.L.; Popp, J. Plasma Proteomic Profiles of Cerebrospinal Fluid-Defined Alzheimer’s Disease Pathology in Older Adults. J. Alzheimer’s Dis. 2017, 60, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Spellman, D.S.; Wildsmith, K.R.; Honigberg, L.A.; Tuefferd, M.; Baker, D.; Raghavan, N.; Nairn, A.C.; Croteau, P.; Schirm, M.; Allard, R.; et al. Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer’s Disease Neuroimaging Initiative (ADNI) CSF. PROTEOMICS—Clin. Appl. 2015, 9, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Vialaret, J.; Schmit, P.-O.; Lehmann, S.; Gabelle, A.; Wood, J.; Bern, M.; Paape, R.; Suckau, D.; Kruppa, G.; Hirtz, C. Identification of multiple proteoforms biomarkers on clinical samples by routine Top-Down approaches. Data Br. 2018, 18, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Gabelle, A.; Vialaret, J.; Schmit, P.-O.; Hirtz, C. Profiling of Intact Proteins in the CSF of Alzheimer’s Disease Patients using Top Down Clinical Proteomics (TDCP): A New Approach Giving Access to Isoform Specific Information of Neurodegenerative Biomarkers. Alzheimer’s Dement. 2016, 12, P183–P184. [Google Scholar] [CrossRef]
- Fania, C.; Arosio, B.; Capitanio, D.; Torretta, E.; Gussago, C.; Ferri, E.; Mari, D.; Gelfi, C. Protein signature in cerebrospinal fluid and serum of Alzheimer’s disease patients: The case of apolipoprotein A-1 proteoforms. PLoS ONE 2017, 12, e0179280. [Google Scholar] [CrossRef] [PubMed]
- Schmit, P.-O.; Vialaret, J.; Wessels, H.J.C.T.; van Gool, A.J.; Lehmann, S.; Gabelle, A.; Wood, J.; Bern, M.; Paape, R.; Suckau, D.; et al. Towards a routine application of Top-Down approaches for label-free discovery workflows. J. Proteom. 2018, 175, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cunningham, R.; Zetterberg, H.; Asthana, S.; Carlsson, C.; Okonkwo, O.; Li, L. Label-free quantitative comparison of cerebrospinal fluid glycoproteins and endogenous peptides in subjects with Alzheimer’s disease, mild cognitive impairment, and healthy individuals. PROTEOMICS—Clin. Appl. 2016, 10, 1225–1241. [Google Scholar] [CrossRef] [PubMed]
- Wildsmith, K.R.; Schauer, S.P.; Smith, A.M.; Arnott, D.; Zhu, Y.; Haznedar, J.; Kaur, S.; Mathews, W.R.; Honigberg, L.A. Identification of longitudinally dynamic biomarkers in Alzheimer’s disease cerebrospinal fluid by targeted proteomics. Mol. Neurodegener. 2014, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Hölttä, M.; Minthon, L.; Hansson, O.; Holmén-Larsson, J.; Pike, I.; Ward, M.; Kuhn, K.; Rüetschi, U.; Zetterberg, H.; Blennow, K.; et al. An Integrated Workflow for Multiplex CSF Proteomics and Peptidomics—Identification of Candidate Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease. J. Proteome Res. 2015, 14, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Hou, S.; Choe, L.H.; Lee, K.H. Targeted human cerebrospinal fluid proteomics for the validation of multiple Alzheimer’s disease biomarker candidates. J. Chromatogr. B 2013, 930, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.-H.; Wu, J.-J.; Ren, H.-M.; Wang, J.; Ding, Z.-T.; Jiang, Y.-P. Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp. Ther. Med. 2016, 11, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; An, J.; Hood, B.L.; Conrads, T.P.; Bowser, R.P. Label-Free LC–MS/MS Proteomic Analysis of Cerebrospinal Fluid Identifies Protein/Pathway Alterations and Candidate Biomarkers for Amyotrophic Lateral Sclerosis. J. Proteome Res. 2015, 14, 4486–4501. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Thézénas, M.-L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Kessler, B.M.; Turner, M.R. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Begcevic, I.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Goldhardt, O.; Grimmer, T.; Magdolen, V.; Batruch, I.; Diamandis, E.P. Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. J. Proteom. 2018, 182, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, R.C.; Lee, A.Y.H.; Song, Q.; Liaw, A.; Wiener, M.; Paweletz, C.P.; Seeburger, J.L.; Li, J.; Meng, F.; Deyanova, E.G.; et al. High Resolution Discovery Proteomics Reveals Candidate Disease Progression Markers of Alzheimer’s Disease in Human Cerebrospinal Fluid. PLoS ONE 2015, 10, e0135365. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Movius, J.; Dator, R.; Aro, P.; Zhao, Y.; Pan, C.; Lin, X.; Bammler, T.K.; Stewart, T.; Zabetian, C.P.; et al. Cerebrospinal fluid peptides as potential Parkinson disease biomarkers: A staged pipeline for discovery and validation. Mol. Cell. Proteom. 2015, 14, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.W.; Heywood, W.E.; Heslegrave, A.J.; Magdalinou, N.K.; Andreasson, U.; Sirka, E.; Bliss, E.; Slattery, C.F.; Toombs, J.; Svensson, J.; et al. A targeted proteomic multiplex CSF assay identifies increased malate dehydrogenase and other neurodegenerative biomarkers in individuals with Alzheimer’s disease pathology. Transl. Psychiatry 2016, 6, e952. [Google Scholar] [CrossRef] [PubMed]
- Magdalinou, N.K.; Noyce, A.J.; Pinto, R.; Lindstrom, E.; Holmén-Larsson, J.; Holtta, M.; Blennow, K.; Morris, H.R.; Skillbäck, T.; Warner, T.T.; et al. Identification of candidate cerebrospinal fluid biomarkers in parkinsonism using quantitative proteomics. Parkinsonism Relat. Disord. 2017, 37, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Brinkmalm, G.; Sjödin, S.; Simonsen, A.H.; Hasselbalch, S.G.; Zetterberg, H.; Brinkmalm, A.; Blennow, K. A Parallel Reaction Monitoring Mass Spectrometric Method for Analysis of Potential CSF Biomarkers for Alzheimer’s Disease. PROTEOMICS—Clin. Appl. 2018, 12, 1700131. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Elias, N.; Koel-Simmelink, M.J.A.; Durieux-Lu, S.; Malekzadeh, A.; Pham, T.V.; Piersma, S.R.; Beccari, T.; Meeter, L.H.H.; Dopper, E.G.P.; et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimer’s Dement. 2016, 2, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Steinacker, P.; von Arnim, C.A.F.; Straub, S.; Nagl, M.; Feneberg, E.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Intact Protein Analysis of Ubiquitin in Cerebrospinal Fluid by Multiple Reaction Monitoring Reveals Differences in Alzheimer’s Disease and Frontotemporal Lobar Degeneration. J. Proteome Res. 2014, 13, 4518–4525. [Google Scholar] [CrossRef] [PubMed]
- Sjödin, S.; Hansson, O.; Öhrfelt, A.; Brinkmalm, G.; Zetterberg, H.; Brinkmalm, A.; Blennow, K. Mass Spectrometric Analysis of Cerebrospinal Fluid Ubiquitin in Alzheimer’s Disease and Parkinsonian Disorders. PROTEOMICS—Clin. Appl. 2017, 11, 1700100. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Bandrowski, A.; Carr, S.; Edwards, A.; Ellenberg, J.; Lundberg, E.; Rimm, D.L.; Rodriguez, H.; Hiltke, T.; Snyder, M.; et al. A proposal for validation of antibodies. Nat. Methods 2016, 13, 823. [Google Scholar] [CrossRef] [PubMed]
- Guglielmo-Viret, V.; Attrée, O.; Blanco-Gros, V.; Thullier, P. Comparison of electrochemiluminescence assay and ELISA for the detection of Clostridium botulinum type B neurotoxin. J. Immunol. Methods 2005, 301, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.S.; Mielke, M.M.; Rosenberg, P.B.; Jain, A.; Fedarko, N.S.; Lyketsos, C.G.; Mehta, P.D. Comparison of conventional ELISA with electrochemiluminescence technology for detection of amyloid-β in plasma. J. Alzheimer’s Dis. 2010, 21, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Swartzman, E.E.; Miraglia, S.J.; Mellentin-Michelotti, J.; Evangelista, L.; Yuan, P.-M. A Homogeneous and Multiplexed Immunoassay for High-Throughput Screening Using Fluorometric Microvolume Assay Technology. Anal. Biochem. 1999, 271, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.B. Development of internal controls for the Luminex instrument as part of a multiplex seven-analyte viral respiratory antibody profile. Clin. Diagn. Lab. Immunol. 2002, 9, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Mustjoki, S.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S.I.; Mangsbo, S.M. The use of multiplex platforms for absolute and relative protein quantification of clinical material. EuPA Open Proteom. 2014, 3, 37–47. [Google Scholar] [CrossRef]
- Kupcova Skalnikova, H.; Cizkova, J.; Cervenka, J.; Vodicka, P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. Int. J. Mol. Sci. 2017, 18, 2697. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011, 39, e102. [Google Scholar] [CrossRef] [PubMed]
- Hensley, P. SOMAmers and SOMAscan—A Protein Biomarker Discovery Platform for Rapid Analysis of Sample Collections From Bench Top to the Clinic. J. Biomol. Tech. 2013, 24, S5. [Google Scholar]
- Dencker, M.; Björgell, O.; Hlebowicz, J. Effect of food intake on 92 neurological biomarkers in plasma. Brain Behav. 2017, 7, e00747. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K. Alzheimer’s Disease Neuroimaging Initiative Association of Plasma Neurofilament Light With Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Breen, E.C.; Reynolds, S.M.; Cox, C.; Jacobson, L.P.; Magpantay, L.; Mulder, C.B.; Dibben, O.; Margolick, J.B.; Bream, J.H.; Sambrano, E.; et al. Multisite comparison of high-sensitivity multiplex cytokine assays. Clin. Vaccine Immunol. 2011, 18, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Muszyński, P.; Groblewska, M.; Kulczyńska-Przybik, A.; Kułakowska, A.; Mroczko, B. YKL-40 as a Potential Biomarker and a Possible Target in Therapeutic Strategies of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Hye, A.; Riddoch-Contreras, J.; Baird, A.L.; Ashton, N.J.; Bazenet, C.; Leung, R.; Westman, E.; Simmons, A.; Dobson, R.; Sattlecker, M.; et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimer's Dement. 2014, 10, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Kiddle, S.J.; Thambisetty, M.; Simmons, A.; Riddoch-Contreras, J.; Hye, A.; Westman, E.; Pike, I.; Ward, M.; Johnston, C.; Lupton, M.K.; et al. Plasma based markers of [11C] PiB-PET brain amyloid burden. PLoS ONE 2012, 7, e44260. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.L.; Westwood, S.; Lovestone, S. Blood-Based Proteomic Biomarkers of Alzheimer’s Disease Pathology. Front. Neurol. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Voyle, N.; Baker, D.; Burnham, S.C.; Covin, A.; Zhang, Z.; Sangurdekar, D.P.; Tan Hehir, C.A.; Bazenet, C.; Lovestone, S.; Kiddle, S.; et al. AIBL research group, and the A. research Blood Protein Markers of Neocortical Amyloid-β Burden: A Candidate Study Using SOMAscan Technology. J. Alzheimer’s Dis. 2015, 46, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, M.; Mollenhauer, B.; Bertolotto, A.; Engelborghs, S.; Hampel, H.; Simonsen, A.H.; Kapaki, E.; Kruse, N.; Le Bastard, N.; Lehmann, S.; et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: An update. Biomark. Med. 2012, 6, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Leitão, M.J.; Baldeiras, I.; Herukka, S.-K.; Pikkarainen, M.; Leinonen, V.; Simonsen, A.H.; Perret-Liaudet, A.; Fourier, A.; Quadrio, I.; Veiga, P.M.; et al. Chasing the Effects of Pre-Analytical Confounders—A Multicenter Study on CSF-AD Biomarkers. Front. Neurol. 2015, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Fourier, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Quadrio, I.; Perret-Liaudet, A. Pre-analytical and analytical factors influencing Alzheimer’s disease cerebrospinal fluid biomarker variability. Clin. Chim. Acta 2015, 449, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Livesey, J.H.; Ellis, M.J.; Evans, M.J. Pre-analytical requirements. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S11–S15. [Google Scholar]
- Le Bastard, N.; De Deyn, P.P.; Engelborghs, S. Importance and Impact of Preanalytical Variables on Alzheimer Disease Biomarker Concentrations in Cerebrospinal Fluid. Clin. Chem. 2015, 61, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, H.M.J.; Janelidze, S.; Demeyer, L.; Coart, E.; Stoops, E.; Herbst, V.; Mauroo, K.; Brix, B.; Hansson, O. Optimized Standard Operating Procedures for the Analysis of Cerebrospinal Fluid Aβ42 and the Ratios of Aβ Isoforms Using Low Protein Binding Tubes. J. Alzheimer’s Dis. 2016, 53, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Vallabh, S.M.; Nobuhara, C.K.; Llorens, F.; Zerr, I.; Parchi, P.; Capellari, S.; Kuhn, E.; Klickstein, J.; Safar, J.; Nery, F.; et al. Prion protein quantification in cerebrospinal fluid as a tool for prion disease drug development. bioRxiv 2018, 295063. [Google Scholar] [CrossRef]
- Comstock, G.W.; Burke, A.E.; Norkus, E.P.; Gordon, G.B.; Hoffman, S.C.; Helzlsouer, K.J. Effects of repeated freeze-thaw cycles on concentrations of cholesterol, micronutrients, and hormones in human plasma and serum. Clin. Chem. 2001, 47, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Jani, D.; Allinson, J.; Berisha, F.; Cowan, K.J.; Devanarayan, V.; Gleason, C.; Jeromin, A.; Keller, S.; Khan, M.U.; Nowatzke, B.; et al. Recommendations for Use and Fit-for-Purpose Validation of Biomarker Multiplex Ligand Binding Assays in Drug Development. AAPS J. 2016, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg-Hasson, Y.; Hansmann, L.; Liedtke, M.; Herschmann, I.; Maecker, H.T. Effects of serum and plasma matrices on multiplex immunoassays. Immunol. Res. 2014, 58, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.; Ward, G. Interferences in immunoassay. Clin. Biochem. Rev. 2004, 25, 105–120. [Google Scholar] [PubMed]
- Martins, T.B.; Pasi, B.M.; Litwin, C.M.; Hill, H.R. Heterophile antibody interference in a multiplexed fluorescent microsphere immunoassay for quantitation of cytokines in human serum. Clin. Diagn. Lab. Immunol. 2004, 11, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Eckels, J.; Schilling, B.; Ludwig, C.; Jaffe, J.D.; MacCoss, M.J.; MacLean, B. Panorama Public: A public repository for quantitative data sets processed in Skyline. Mol. Cell. Proteom. 2018, 17, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Khoonsari, P.E.; Häggmark, A.; Lönnberg, M.; Mikus, M.; Kilander, L.; Lannfelt, L.; Bergquist, J.; Ingelsson, M.; Nilsson, P.; Kultima, K.; et al. Analysis of the Cerebrospinal Fluid Proteome in Alzheimer’s Disease. PLoS ONE 2016, 11, e0150672. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Andrews, T.D.; Bevan, P.; Caccamo, M.; Chen, Y.; Clarke, L.; Coates, G.; Cuff, J.; Curwen, V.; Cutts, T.; et al. An overview of Ensembl. Genome Res. 2004, 14, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Guldbrandsen, A.; Farag, Y.; Kroksveen, A.C.; Oveland, E.; Lereim, R.R.; Opsahl, J.A.; Myhr, K.-M.; Berven, F.S.; Barsnes, H. CSF-PR 2.0: An Interactive Literature Guide to Quantitative Cerebrospinal Fluid Mass Spectrometry Data from Neurodegenerative Disorders. Mol. Cell. Proteom. 2017, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]



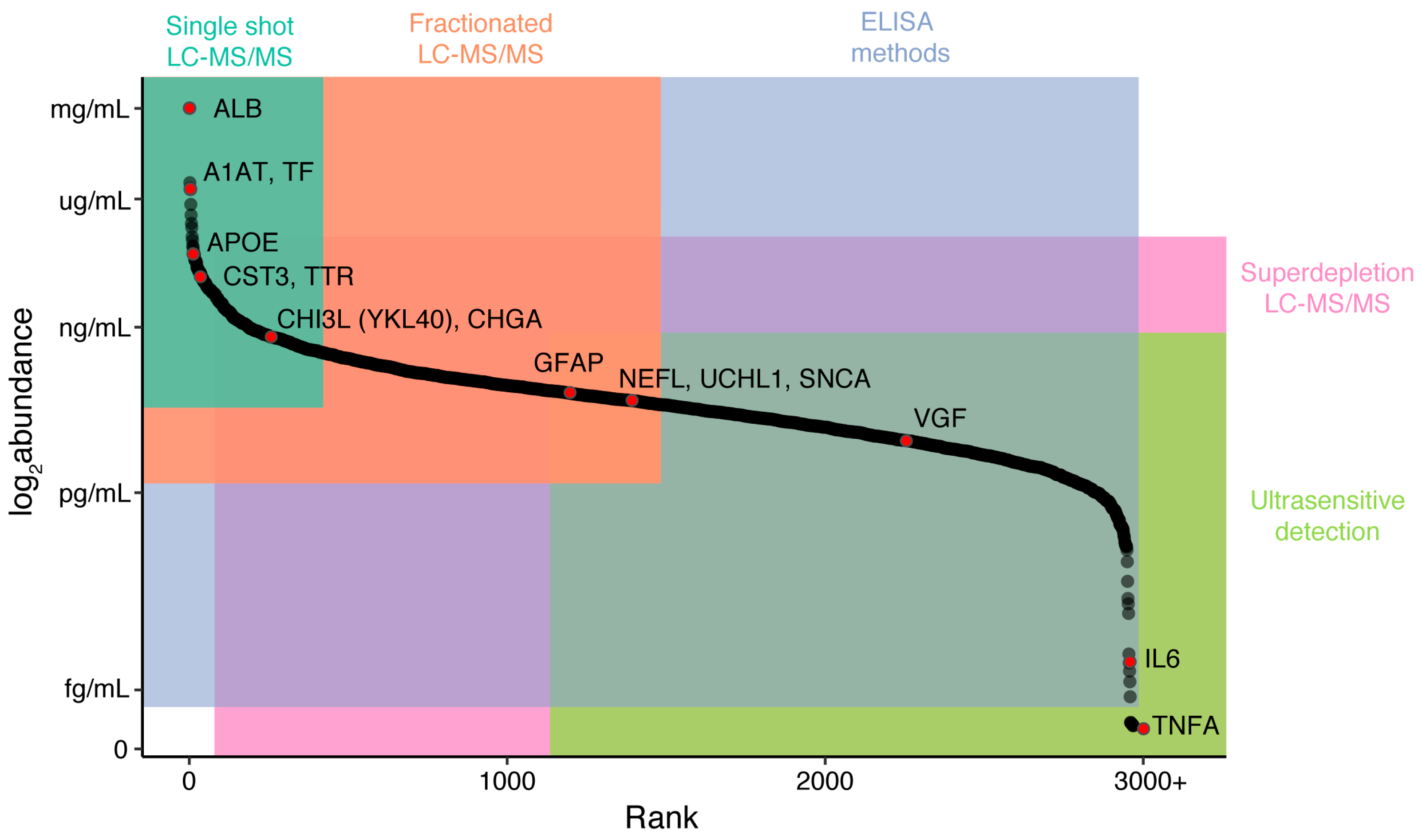{kind=link}
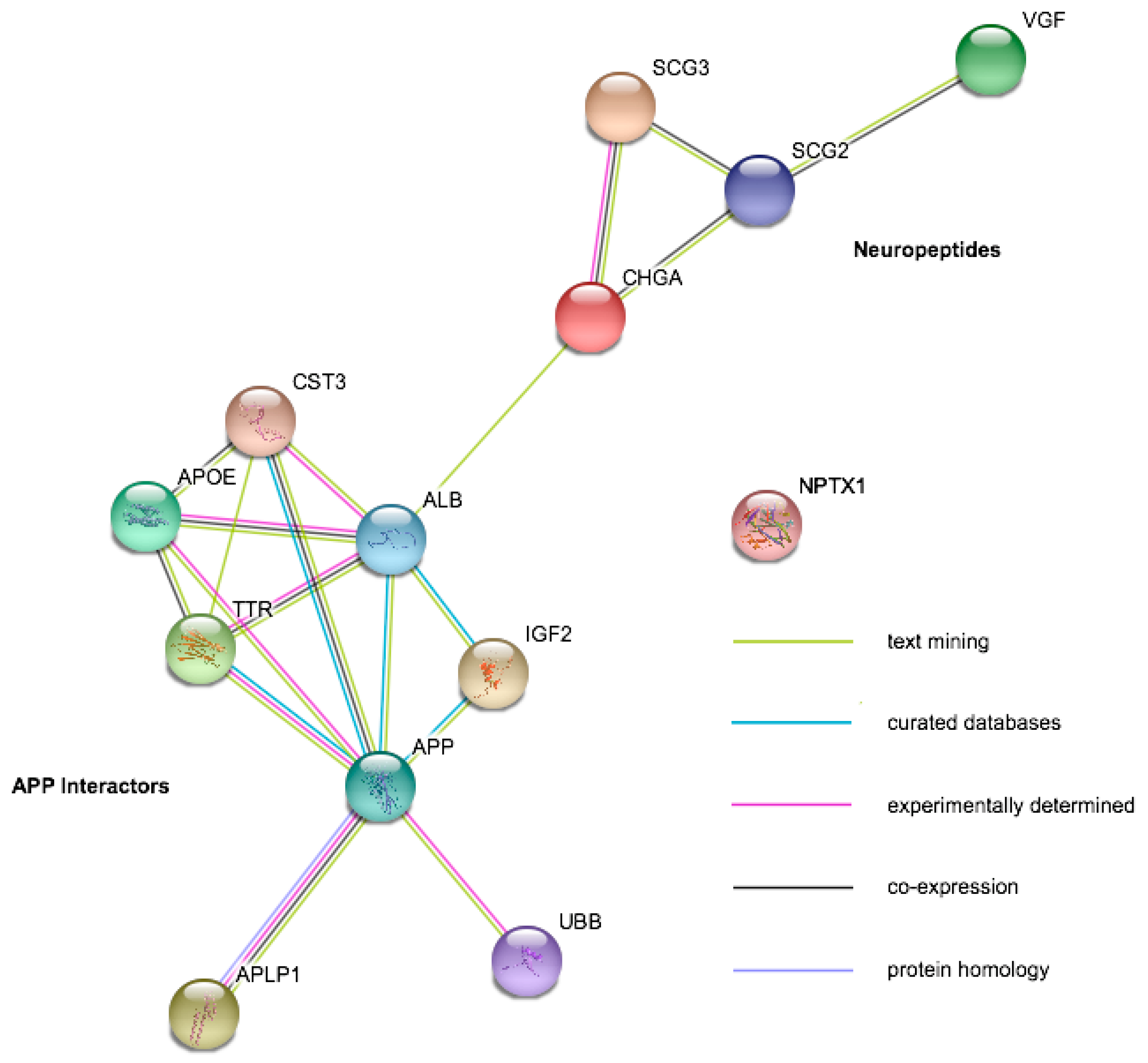{kind=link}
| Protein | Gene Symbol | Mild Cognitive Impairment | Alzheimer’s Disease | Amyotrophic Lateral Sclerosis | Other Diseases |
|---|---|---|---|---|---|
| Serum albumin | ALB | ↔ [89,90] | ↓ [48,91,92] ↑ [48,92] ↔ [89,90] | ↔ [93,94,95] | |
| Amyloid Beta Precursor Like Protein | APLP1 | ↑ [96] ↔ [89,90] | ↔ [89,90,96,97,98] ↓ [91] ↑ [98] | ↔ [94,95] | ↓ PD [98] |
| Apolipoprotein E | APOE | ↓ [89] ↔ [90] | ↑ [48,92,99,100] ↔ [83,90,91,97] ↓ [89] | ↔ [93,94,95] | ↔ PD [98,99] ↑ LBD [99] |
| Amyloid Precursor Protein | APP | ↔ [90] | ↔ [83,89,90,96] ↓ [97] | ↔ [93,94,95] | ↔ PD [98,99] ↑ LBD [99] ↓ APS [101] |
| Chromogranin A | CHGA | ↔ [89,90] | ↓ [91,97,102] ↔ [89,90,92] | ↔ [93,94,95] | |
| Chitinase 3 Like 1 (YKL-40) | CHI3L | ↔ [89,90] | ↑ [90,99,100] ↔ [83,89] | ↔ [93,94] ↑ [95] | ↔ PD [99] ↑ LBD [99] ↑ FTD [103] ↑ APS [101] |
| Cystatin-C | CST3 | ↔ [89,90] | ↓ [102] ↑ [92,99,100] ↔ [89,90,91,97] | ↔ [93,95] ↓ [94] | ↔ PD [98,99] ↑ LBD [99] |
| Insulin Like Growth Factor-2 | IGF2 | ↔ [89] | ↑ [99,100] ↔ [89] | ↓ [93] ↔ [95] | ↔ PD [99] ↑ LBD [99] |
| Neuronal Pentraxin 1 | NPTX1 | ↓ [89] ↔ [96] | ↓ [89,102] ↔ [83,96] | ↔ [93,94,95] | ↔ PD [98] ↓ APS [101] |
| Secretogranin-2 | SCG2 | ↔ [96] | ↓ [91,102] ↔ [83,96] | ↔ [93,95] ↓ [94] | ↓ APS [101] |
| Secretogranin-3 | SCG3 | ↔ [89,96] | ↔ [83,89,96] ↓ [91,97] ↑ [48] | ↔ [93,94,95] | ↓ APS [101] |
| Transthyretin | TTR | ↑ [89,90] | ↑ [90,92,99] ↔ [83,91,97,100] | ↔ [93,94] | ↔ PD [99] ↔ LBD [99] |
| Ubiquitin (mono/poly) | UBB | ↑ [48,99,104,105] ↔ [83] | ↔ [94,95,104] | ↔ FTD [104] ↔ APS [105] ↑ LBD [99] ↔ PD [99,104,105] | |
| Neurosecretory Protein VGF | VGF | ↔ [89,96] | ↓ [91,97,102] ↔ [83,89,96] | ↔ [93,94,95] | ↓ APS [101] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlyle, B.C.; Trombetta, B.A.; Arnold, S.E. Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias. Proteomes 2018, 6, 32. https://doi.org/10.3390/proteomes6030032
Carlyle BC, Trombetta BA, Arnold SE. Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias. Proteomes. 2018; 6(3):32. https://doi.org/10.3390/proteomes6030032
Chicago/Turabian StyleCarlyle, Becky C., Bianca A. Trombetta, and Steven E. Arnold. 2018. "Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias" Proteomes 6, no. 3: 32. https://doi.org/10.3390/proteomes6030032
APA StyleCarlyle, B. C., Trombetta, B. A., & Arnold, S. E. (2018). Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias. Proteomes, 6(3), 32. https://doi.org/10.3390/proteomes6030032