Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls



Abstract
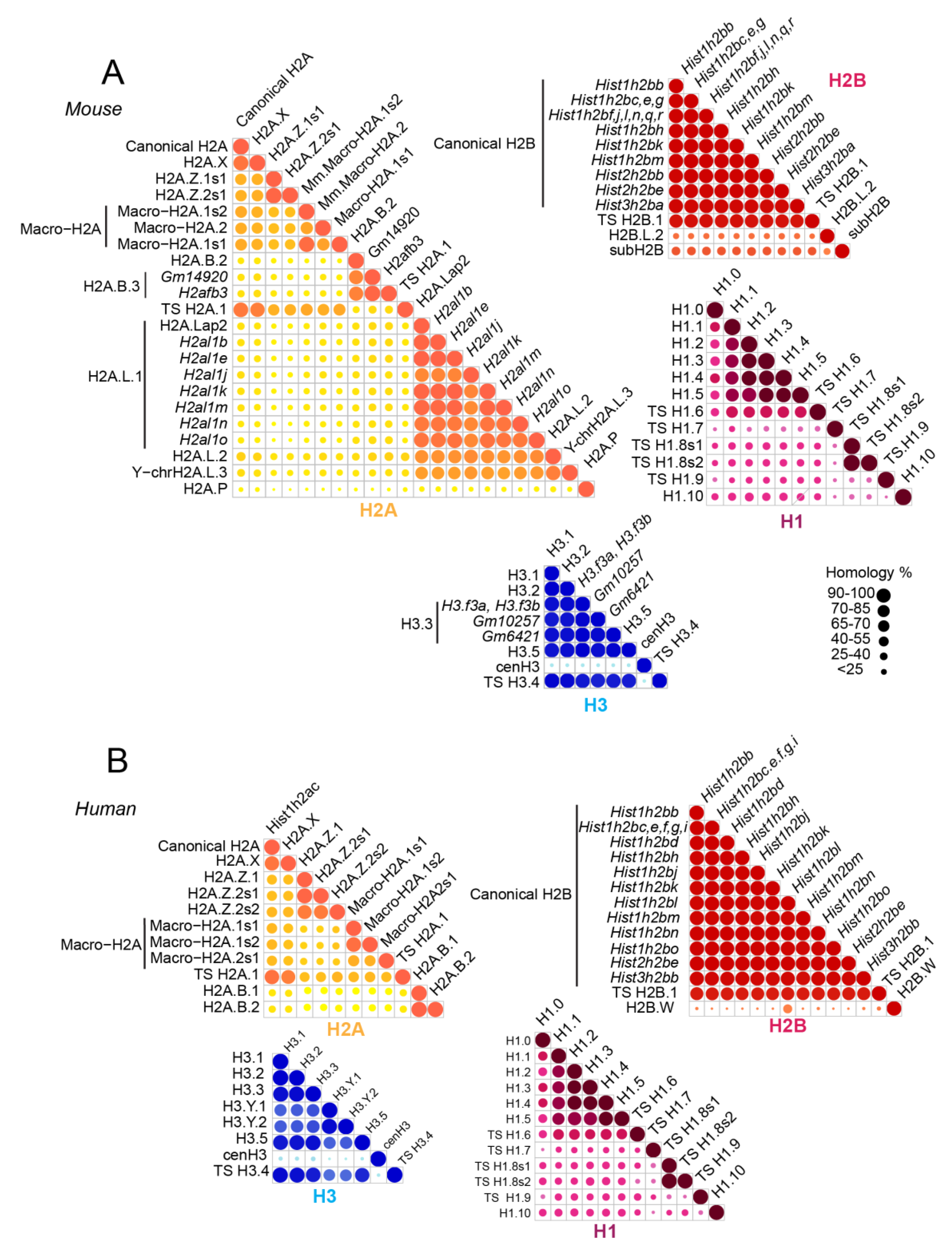
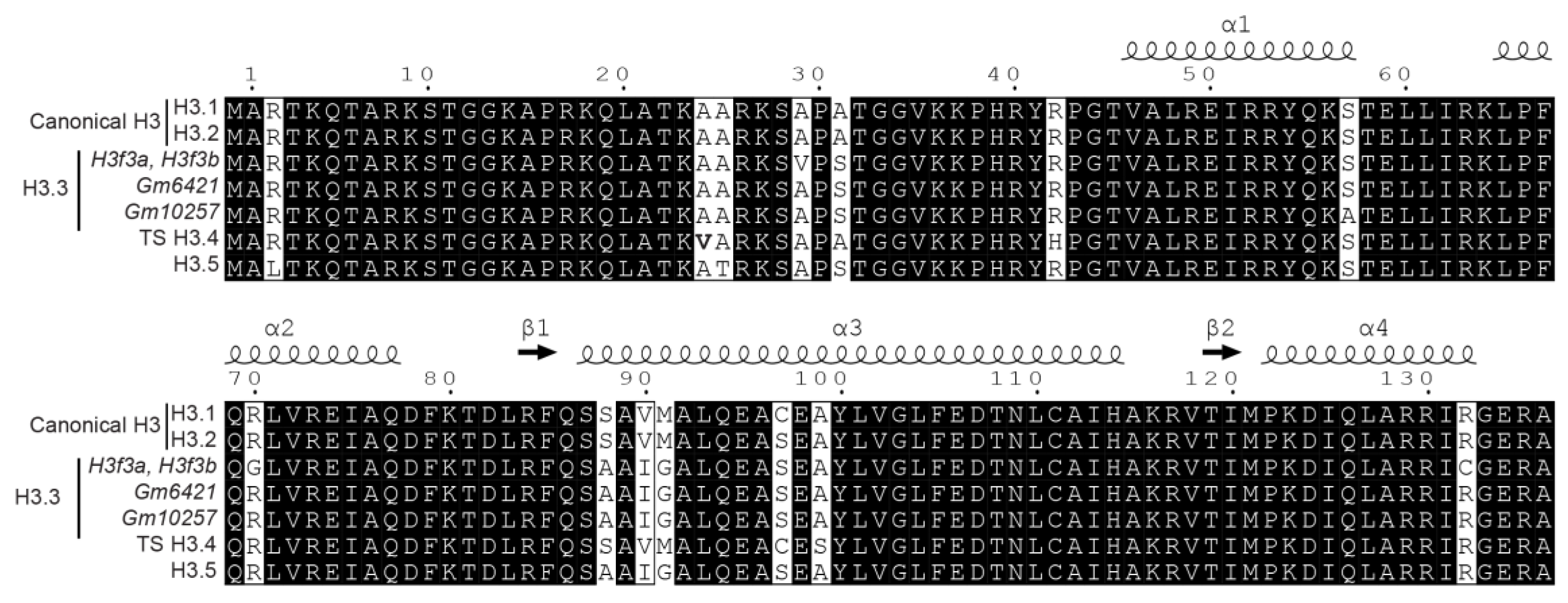
1. Introduction
2. Extracting Histones from Biological Samples
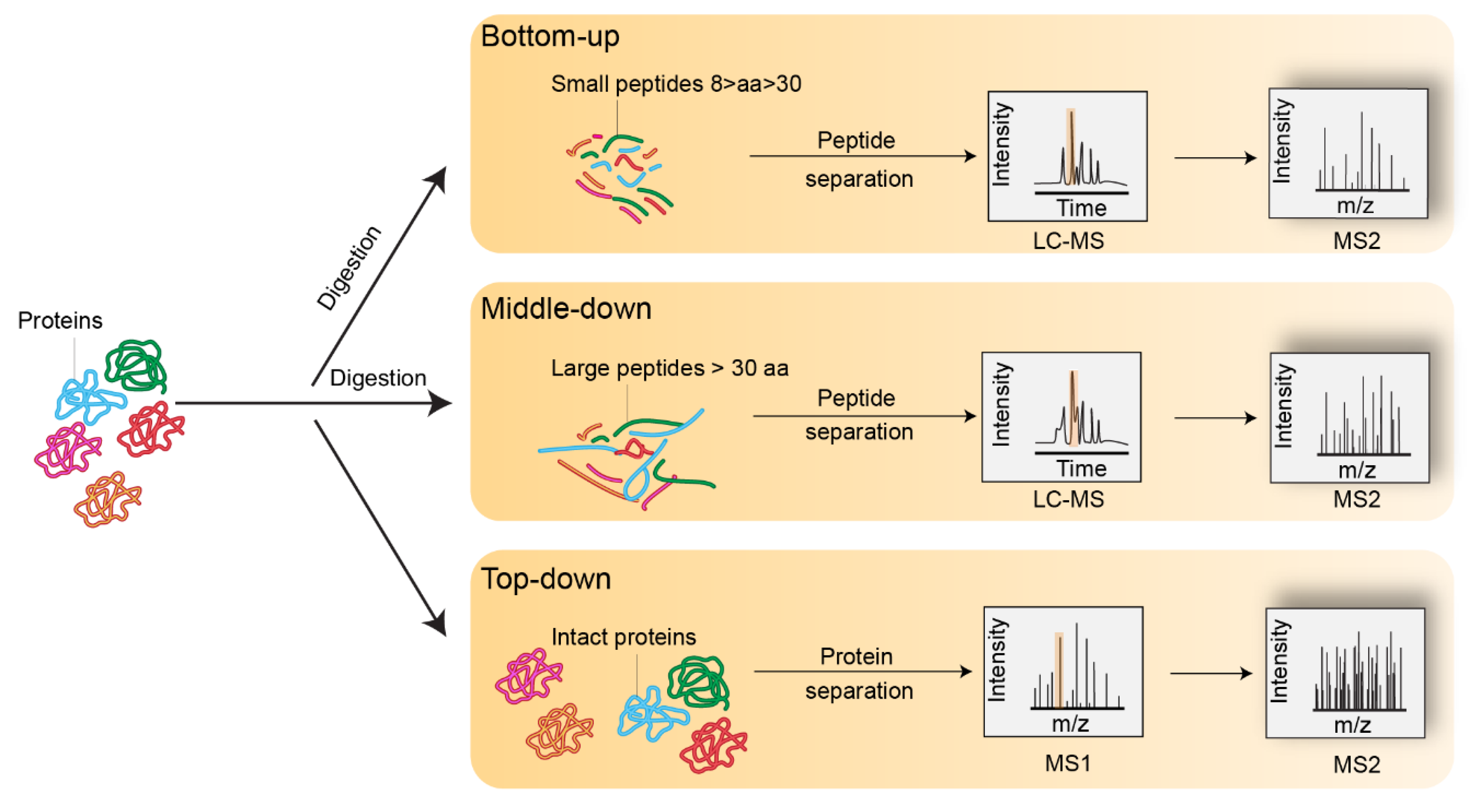
3. Bottom-Up Mass Spectrometry Analysis of Histones
4. Data Interpretation
5. The Puzzle of Histone PTMs
5.1. Frequent Existence of Strictly Isobaric Peptides
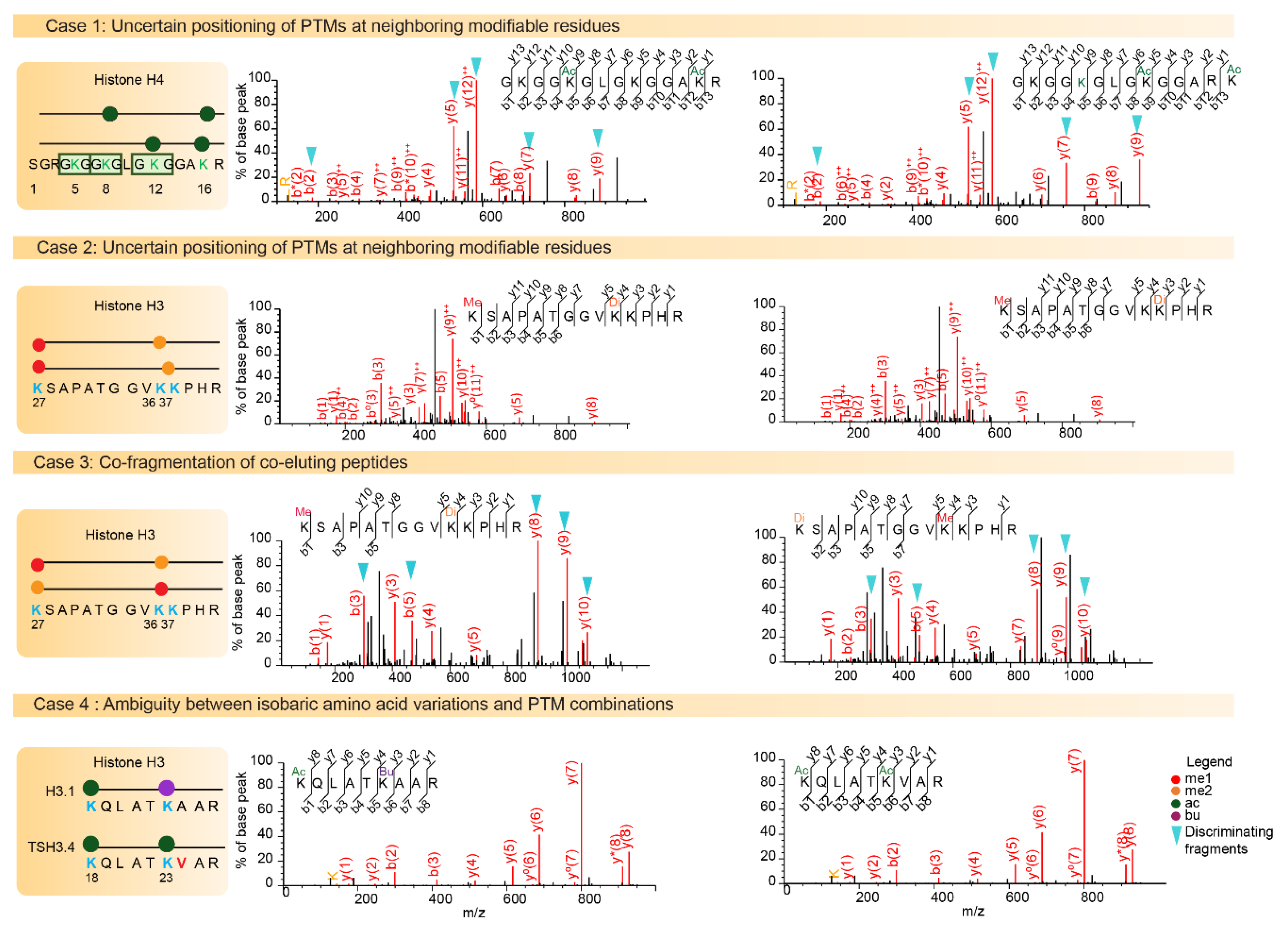
5.2. Uncertain Positioning of PTMs on Neighboring Modifiable Residues
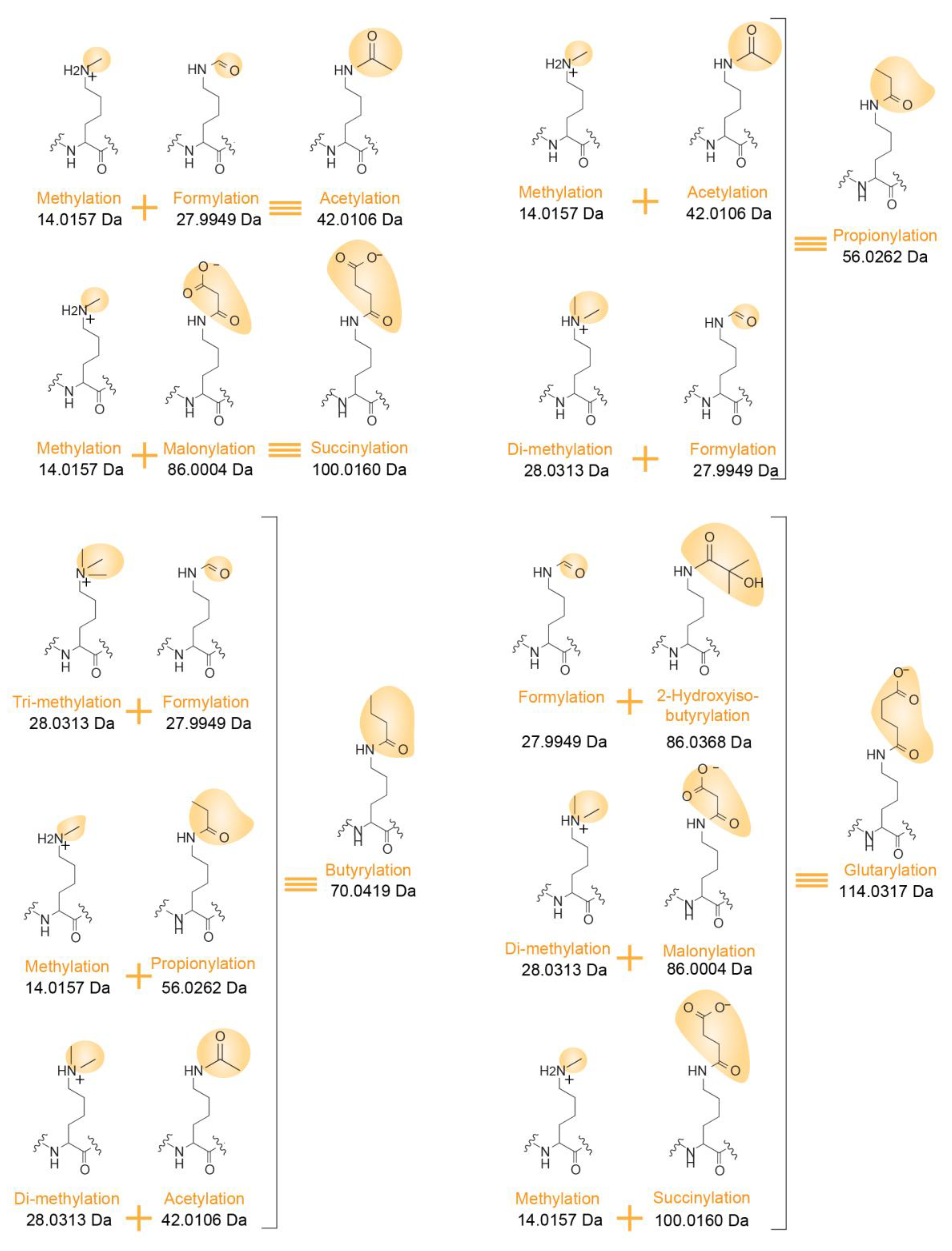
5.3. Ambiguity between Several Isobaric PTM Combinations
5.4. Distinguishing between Isobaric Amino Acid Variations and PTM Combinations
6. Positional Isomer Quantification
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A.; Pałyga, J. Linker histone subtypes and their allelic variants. Cell Biol. Int. 2012, 36, 981–996. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Bednar, J.; Garcia-Saez, I.; Boopathi, R.; Cutter, A.R.; Papai, G.; Reymer, A.; Syed, S.H.; Lone, I.N.; Tonchev, O.; Crucifix, C.; et al. Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 2017, 66, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Schneider, R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Torres-Padilla, M.-E. Developmental biology: Panoramic views of the early epigenome. Nature 2016, 537, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Quenet, D. Histone Variants and Disease. Int. Rev. Cell Mol. Biol. 2018, 335, 1–39. [Google Scholar] [CrossRef] [PubMed]
- El Kennani, S.; Adrait, A.; Shaytan, A.K.; Khochbin, S.; Bruley, C.; Panchenko, A.R.; Landsman, D.; Pflieger, D.; Govin, J. MS_HistoneDB, a manually curated resource for proteomic analysis of human and mouse histones. Epigenet. Chromatin 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Banaszynski, L.A.; Allis, C.D.; Lewis, P.W. Histone Variants in Metazoan Development. Dev. Cell 2010, 19, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Henikoff, S. Histone variants: Dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Bonisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- Hoghoughi, N.; Barral, S.; Vargas, A.; Rousseaux, S.; Khochbin, S. Histone variants: Essential actors in male genome programming. J. Biochem. 2018, 163, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Montellier, E.; Boussouar, F.; Rousseaux, S.; Zhang, K.; Buchou, T.; Fenaille, F.; Shiota, H.; Debernardi, A.; Hery, P.; Curtet, S.; et al. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev. 2013, 27, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- El Kennani, S.; Adrait, A.; Permiakova, O.; Hesse, A.-M.; Ialy-Radio, C.; Ferro, M.; Brun, V.; Cocquet, J.; Govin, J.; Pflieger, D. Systematic quantitative analysis of H2A and H2B variants by targeted proteomics. Epigenet. Chromatin 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Peach, S.E.; Rudomin, E.L.; Udeshi, N.D.; Carr, S.A.; Jaffe, J.D. Quantitative assessment of chromatin immunoprecipitation grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol. Cell. Proteom. 2012, 11, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine Propionylation and Butyrylation Are Novel Post-translational Modifications in Histones. Mol. Cell. Proteom. 2007, 6, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine Succinylation and Lysine Malonylation in Histones. Mol. Cell. Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Peng, C.; Montellier, E.; Lu, Z.; Chen, Y.; Ishii, H.; Debernardi, A.; Buchou, T.; Rousseaux, S.; Jin, F.; et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014, 10, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Bhanu, N.V.; Karch, K.R.; Wang, X.; Garcia, B.A. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, 54112. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Pentakota, S.; Mishra, L.N.; Jones, R.; Rao, M.R.S. Chapter Seven—Identification of Posttranslational Modifications of Endogenous Chromatin Proteins From Testicular Cells by Mass Spectrometry. In Proteomics in Biology, Part B; Shukla, A.K., Ed.; Methods in Enzymology; Academic Press: New York, NY, USA, 2017; Volume 586, pp. 115–142. [Google Scholar]
- Sidoli, S.; Garcia, B.A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteom. 2017, 14, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.; Thompson, P.; Hofseth, L. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Shabanowitz, J.; Hunt, D.F. Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2007, 11, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, A.; Zhang, D.; Huang, H.; Barral, S.; Kwon, O.K.; Qi, S.; Tang, Z.; Buchou, T.; Vitte, A.-L.; He, T.; et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol. Cell 2016, 62, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausió, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.L.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenet. Chromatin 2012, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Draizen, E.J.; Shaytan, A.K.; Mariño-Ramírez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A histone database with variants—An integrated resource to explore histones and their variants. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-F.; Lin, S.; Molden, R.C.; Garcia, B.A. Evaluation of proteomic search engines for the analysis of histone modifications. J. Proteome Res. 2014, 13, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, W.; Cobb, M.H.; Zhao, Y. PTMap—A sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. USA 2009, 106, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, Y.; Zhang, Z.; Zhao, Y. Identification and Verification of Lysine Propionylation and Butyrylation in Yeast Core Histones Using PTMap Software. J. Proteome Res. 2009, 8, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, J.J.; Bullock, C.R.; LeDuc, R.D.; Mizzen, C.A.; Kelleher, N.L. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J. Biol. Chem. 2008, 283, 14927–14937. [Google Scholar] [CrossRef] [PubMed]
- Feller, C.; Forné, I.; Imhof, A.; Becker, P.B. Global and Specific Responses of the Histone Acetylome to Systematic Perturbation. Mol. Cell 2015, 57, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Shuai, L.; Zhu, M.; Liu, P.; Xie, Z.-F.; Jiang, S.; Jiang, H.-W.; Li, J.; Zhao, Y.; Li, J.-Y.; et al. The Landscape of Histone Modifications in a High-Fat Diet-Induced Obese (DIO) Mouse Model. Mol. Cell. Proteom. 2017, 16, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-G.; Suzuki, T.; Dohmae, N. Global mapping of post-translational modifications on histone H3 variants in mouse testes. Biochem. Biophys. Rep. 2017, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Macek, B.; Lange, O.; Makarov, A.; Horning, S.; Mann, M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, C.D.; Frese, C.; Heck, A.J.R.; Olsen, J.V.; Nielsen, M.L. Analytical Utility of Mass Spectral Binning in Proteomic Experiments by SPectral Immonium Ion Detection (SPIID). Mol. Cell. Proteom. 2014, 13, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.R.; Unwin, R.D.; Evans, C.A.; Leech, S.H.; Corfe, B.M.; Whetton, A.D. The Application of a Hypothesis-driven Strategy to the Sensitive Detection and Location of Acetylated Lysine Residues. J. Am. Soc. Mass Spectrom. 2007, 18, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.A.; Ow, S.Y.; Smith, D.L.; Corfe, B.M.; Wright, P.C. Application of the CIRAD Mass Spectrometry Approach for Lysine Acetylation Site Discovery. In Protein Acetylation: Methods and Protocols; Hake, S.B., Janzen, C.J., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 13–23. ISBN 978-1-62703-305-3. [Google Scholar]
- Karch, K.R.; Zee, B.M.; Garcia, B.A. High resolution is not a strict requirement for characterization and quantification of histone post-translational modifications. J. Proteome Res. 2014, 13, 6152–6159. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-G.; Dohmae, N. Proteomic characterization of histone variants in the mouse testis by mass spectrometry-based top-down analysis. Biosci. Trends 2016, 10, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yau, P.M.; Chandrasekhar, B.; New, R.; Kondrat, R.; Imai, B.S.; Bradbury, M.E. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: An application for determining lysine 9 acetylation and methylation of histone H3. Proteomics 2004, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Couttas, T.A.; Raftery, M.J.; Bernardini, G.; Wilkins, M.R. Immonium Ion Scanning for the Discovery of Post-Translational Modifications and Its Application to Histones. J. Proteome Res. 2008, 7, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.; Satomi, Y.; Yoshikawa, K.; Takao, T. Epsilon-N,N,N-trimethyllysine-specific ions in matrix-assisted laser desorption/ionization-tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.A.; Hamilton, B.R.; Wallis, T.P.; Furness, S.G.B.; Whitelaw, M.L.; Gorman, J.J. Identification of N,Nε-dimethyl-lysine in the murine dioxin receptor using MALDI-TOF/TOF- and ESI-LTQ-Orbitrap-FT-MS. Int. J. Mass Spectrom. 2007, 268, 168–180. [Google Scholar] [CrossRef]
- Snijders, A.P.L.; Hung, M.-L.; Wilson, S.A.; Dickman, M.J. Analysis of arginine and lysine methylation utilizing peptide separations at neutral pH and electron transfer dissociation mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Brame, C.J.; Moran, M.F.; McBroom-Cerajewski, L.D.B. A mass spectrometry based method for distinguishing between symmetrically and asymmetrically dimethylated arginine residues. Rapid Commun. Mass Spectrom. 2004, 18, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Straubinger, R.M.; Aletta, J.M.; Cao, J.; Duan, X.; Yu, H.; Qu, J. Accurate localization and relative quantification of arginine methylation using nanoflow liquid chromatography coupled to electron transfer dissociation and orbitrap mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 2004, 1, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Friesen, W.J.; Paushkin, S.; Dreyfuss, G.; Mann, M. Detection of Arginine Dimethylated Peptides by Parallel Precursor Ion Scanning Mass Spectrometry in Positive Ion Mode. Anal. Chem. 2003, 75, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, P.M.; Hunziker, P.E.; Zahariev, S.; Pongor, S. Fragmentation pathways of NG-methylated and unmodified arginine residues in peptides studied by ESI-MS/MS and MALDI-MS. J. Am. Soc. Mass Spectrom. 2004, 15, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chen, J.; Gao, Y.; Gao, J.; Liao, R.; Wang, Y.; Oyang, C.; Li, E.; Zeng, C.; Zhou, S.; et al. Quantitative Profiling of Combinational K27/K36 Modifications on Histone H3 Variants in Mouse Organs. J. Proteome Res. 2016, 15, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Darwanto, A.; Curtis, M.P.; Schrag, M.; Kirsch, W.; Liu, P.; Xu, G.; Neidigh, J.W.; Zhang, K. A modified “cross-talk” between histone H2B Lys-120 ubiquitination and H3 Lys-79 methylation. J. Biol. Chem. 2010, 285, 21868–21876. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.C.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-F.; Lin, S.; Molden, R.C.; Cao, X.-J.; Bhanu, N.V.; Wang, X.; Sidoli, S.; Liu, S.; Garcia, B.A. EpiProfile quantifies histone modifications by extracting retention time and intensity in high-resolution mass spectra. Mol. Cell. Proteom. 2015, 14, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Lin, S.; Xiong, L.; Bhanu, N.V.; Karch, K.R.; Johansen, E.; Hunter, C.; Mollah, S.; Garcia, B.A. Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol. Cell. Proteom. 2015, 14, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Simithy, J.; Karch, K.R.; Kulej, K.; Garcia, B.A. Low Resolution Data-Independent Acquisition in an LTQ-Orbitrap Allows for Simplified and Fully Untargeted Analysis of Histone Modifications. Anal. Chem. 2015, 87, 11448–11454. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Reiter, L.; Denu, J.M.; Dowell, J.A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J. Proteome Res. 2015, 14, 3252–3262. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Fujiwara, R.; Kulej, K.; Garcia, B.A. Differential quantification of isobaric phosphopeptides using data-independent acquisition mass spectrometry. Mol. Biosyst. 2016, 12, 2385–2388. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Residues | PTMs | Diagnostic Ion Type | m/z | MS Fragmentation | References |
|---|---|---|---|---|---|
| Lysine | Acetylation | IM-NH3 | 126.091 | CID, HCD | [48,49,50,51] |
| Methylation | IM-NH3 | 98.0964 | CID, HCD | [52,53] | |
| Dimethylation | IM | 112.1 | CID, HCD | [52,53,54] | |
| Trimethylation | NL | 59.0735 | CID | [51,52,53,54] | |
| Propionyl | IM | 140.106 | CID | [45] | |
| Crotonyl | __ | __ | __ | __ | |
| Butyryl | IM | 154.123 | HCD | [45] | |
| Malonyl | __ | __ | __ | __ | |
| 2-hydroxyisobutyryl | __ | __ | __ | __ | |
| Succinyl | __ | __ | __ | __ | |
| Gluratyl | __ | __ | __ | __ | |
| Formyl | IM | 112.0756 | HCD | [47,51,55] | |
| Arginine | Methylation | NL | 73.064 | ETD | [52,53,54,56,57,58] |
| Methylation | NL | 31.0422 | ETD | [52,53,54,56,57,58] | |
| Dimethylation | SC | 46.0651 | CID | [59,60] | |
| Dimethylation-symmetric/asymmetric | SC | 71.0604 | CID | [59,60] | |
| Dimethyl-symmetric | NL | 31.0417 | CID | [56,57,61] | |
| Dimethylation-symmetric | NL | 46.0651 | CID | 59,60,64] | |
| Dimethylation-asymmetric | NL | 45.0573 | CID | [56,57,61] | |
| Tyrosine | phosphoryl | NL | 216.0426 | CID | [47,62] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Kennani, S.; Crespo, M.; Govin, J.; Pflieger, D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes 2018, 6, 29. https://doi.org/10.3390/proteomes6030029
El Kennani S, Crespo M, Govin J, Pflieger D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes. 2018; 6(3):29. https://doi.org/10.3390/proteomes6030029
Chicago/Turabian StyleEl Kennani, Sara, Marion Crespo, Jérôme Govin, and Delphine Pflieger. 2018. "Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls" Proteomes 6, no. 3: 29. https://doi.org/10.3390/proteomes6030029
APA StyleEl Kennani, S., Crespo, M., Govin, J., & Pflieger, D. (2018). Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes, 6(3), 29. https://doi.org/10.3390/proteomes6030029