Proteome Profiling of Diabetic Mellitus Patient Urine for Discovery of Biomarkers by Comprehensive MS-Based Proteomics


Abstract
:1. Introduction
2. Materials and Methods
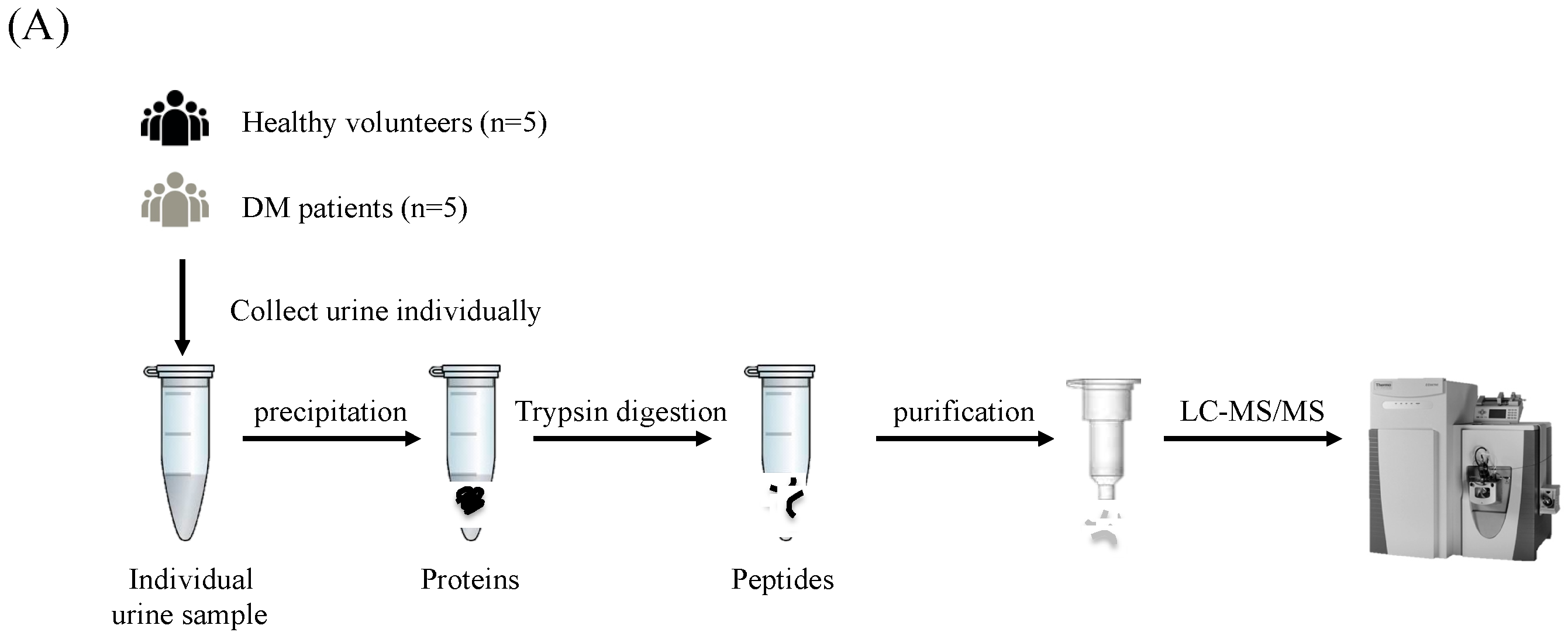
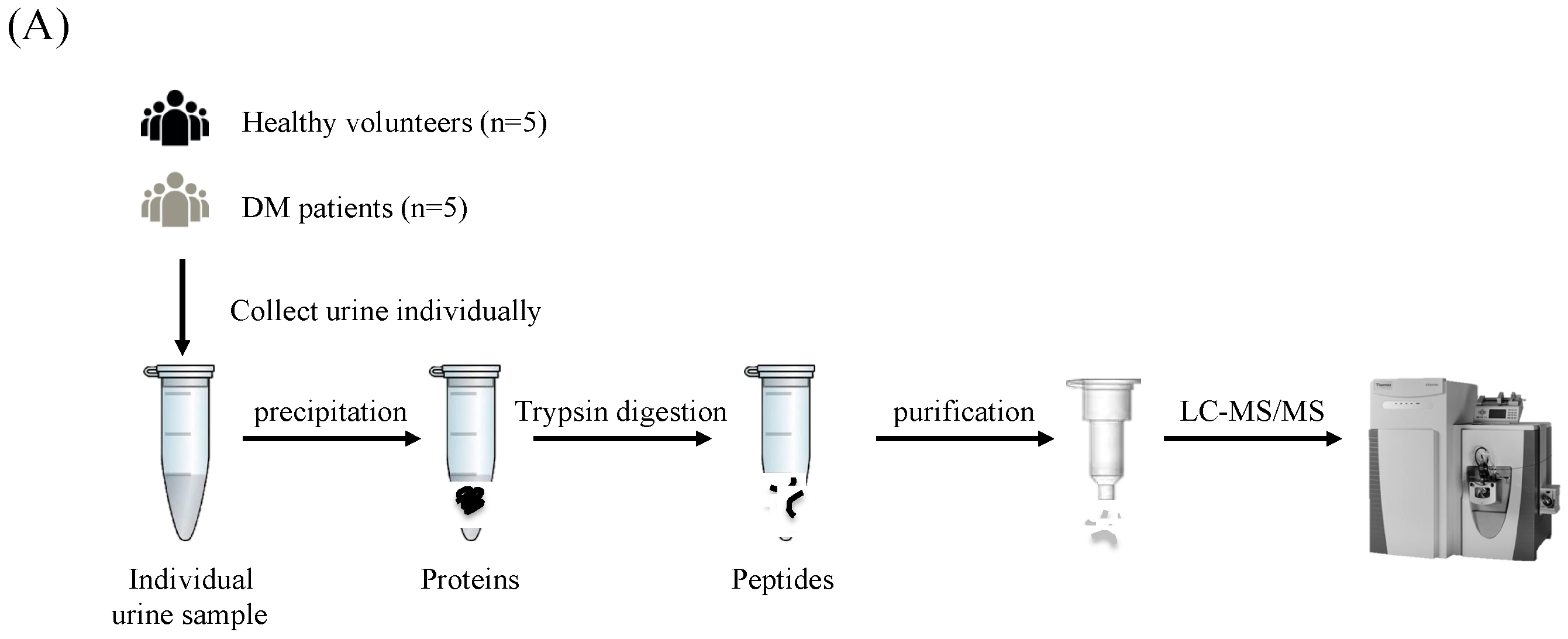
2.1. Urine Samples
2.2. Urine Protein Preparation
2.3. Tryptic Peptide Preparation
2.4. LC-MS/MS Analysis and Protein Identification
3. Results
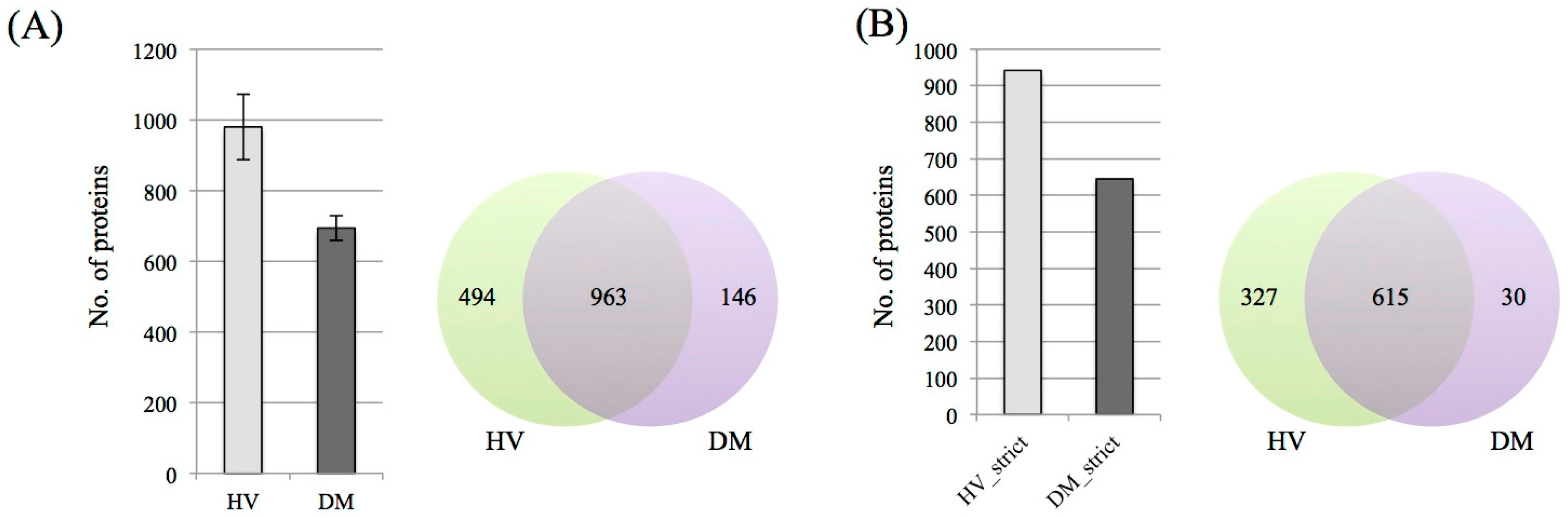
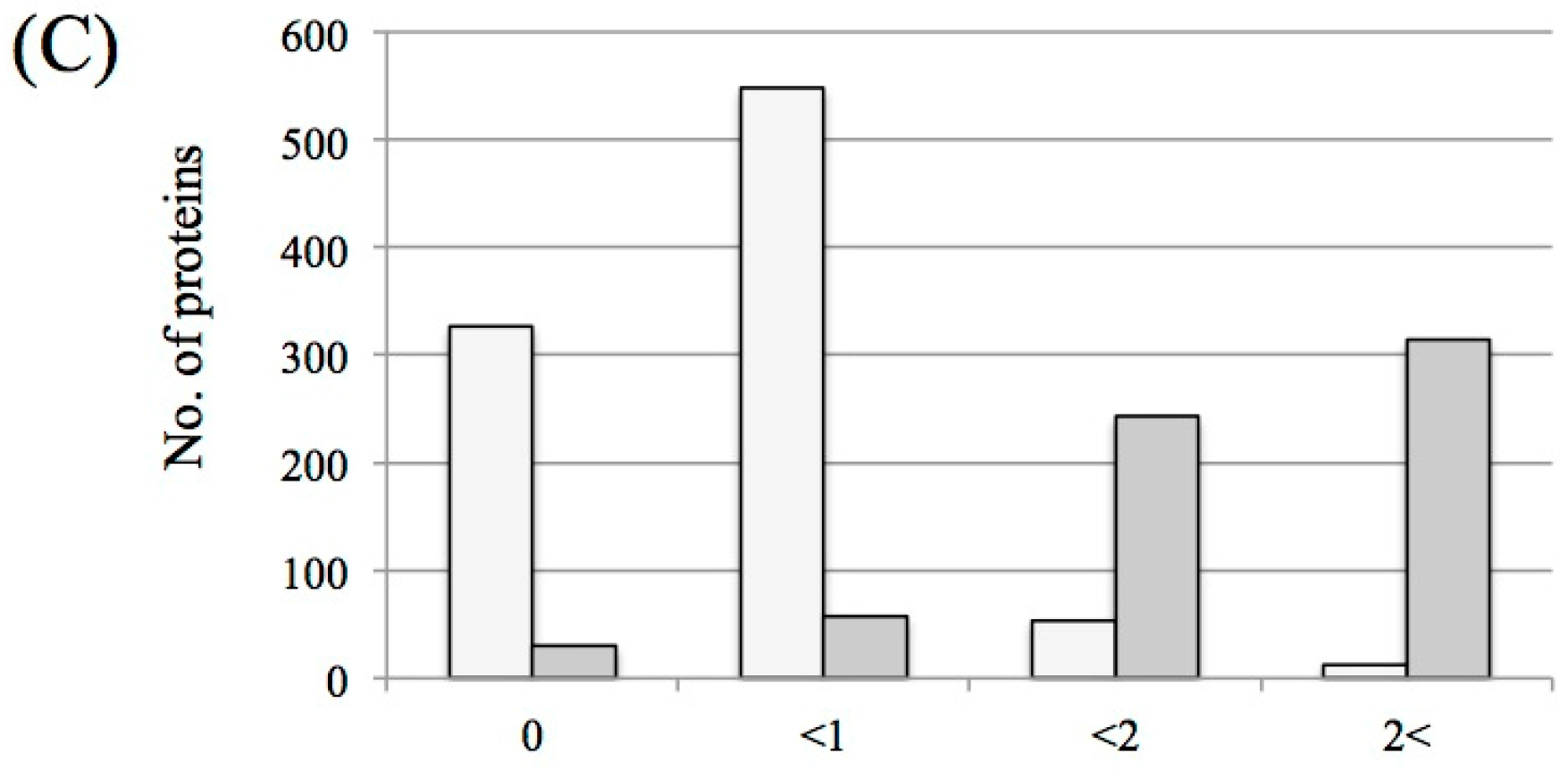
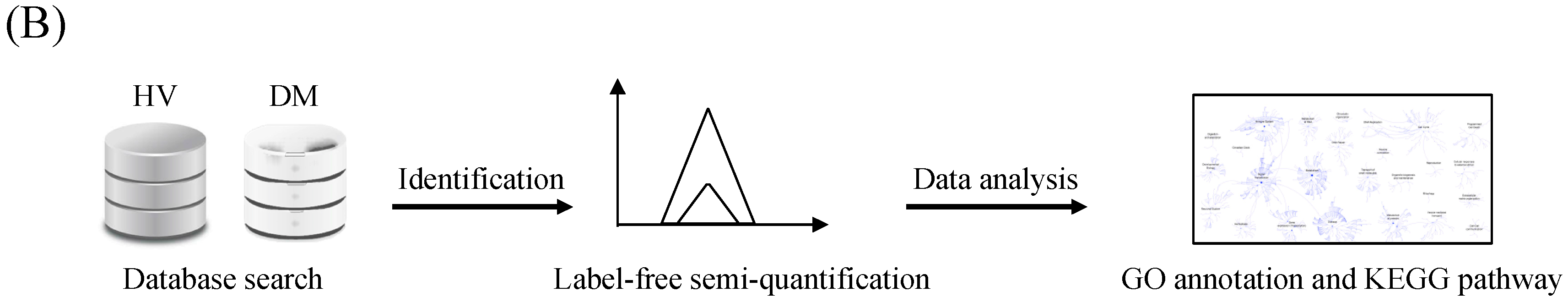
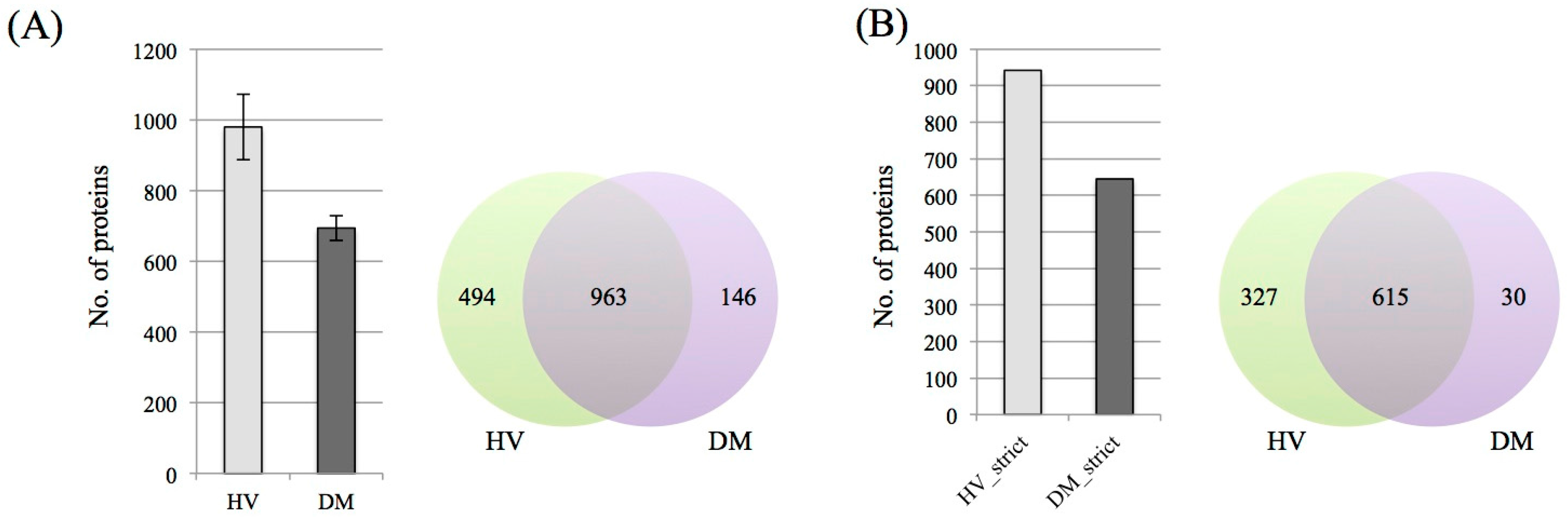
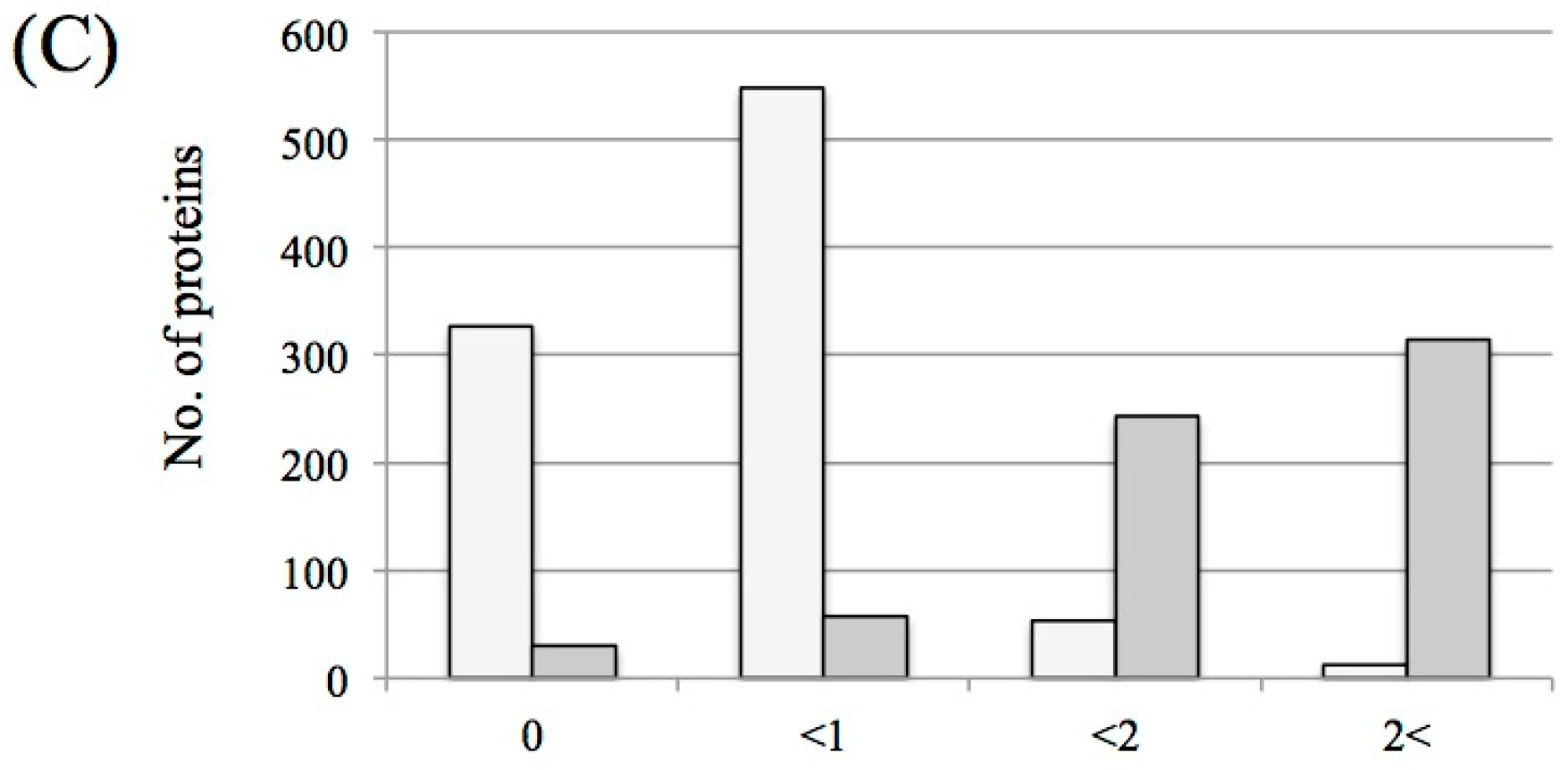
3.1. Protein Identification and Label-Free Semi-Quantification
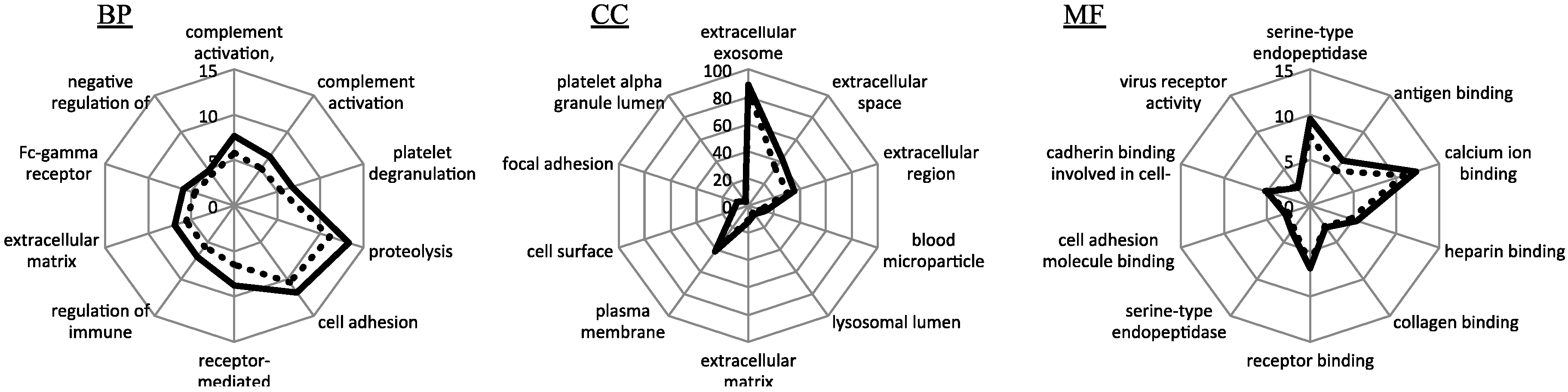
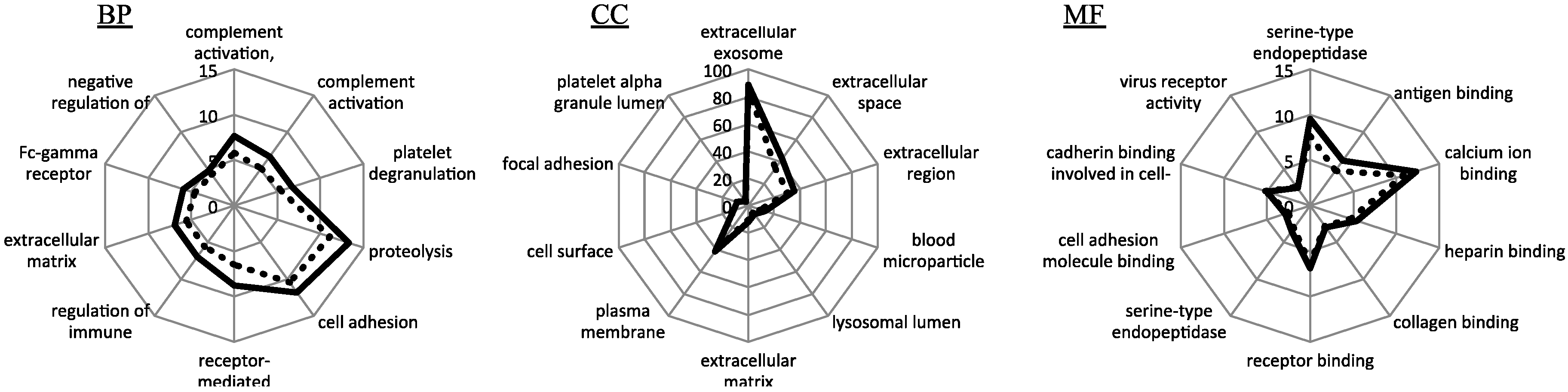
3.2. Functional Enrichment Analysis
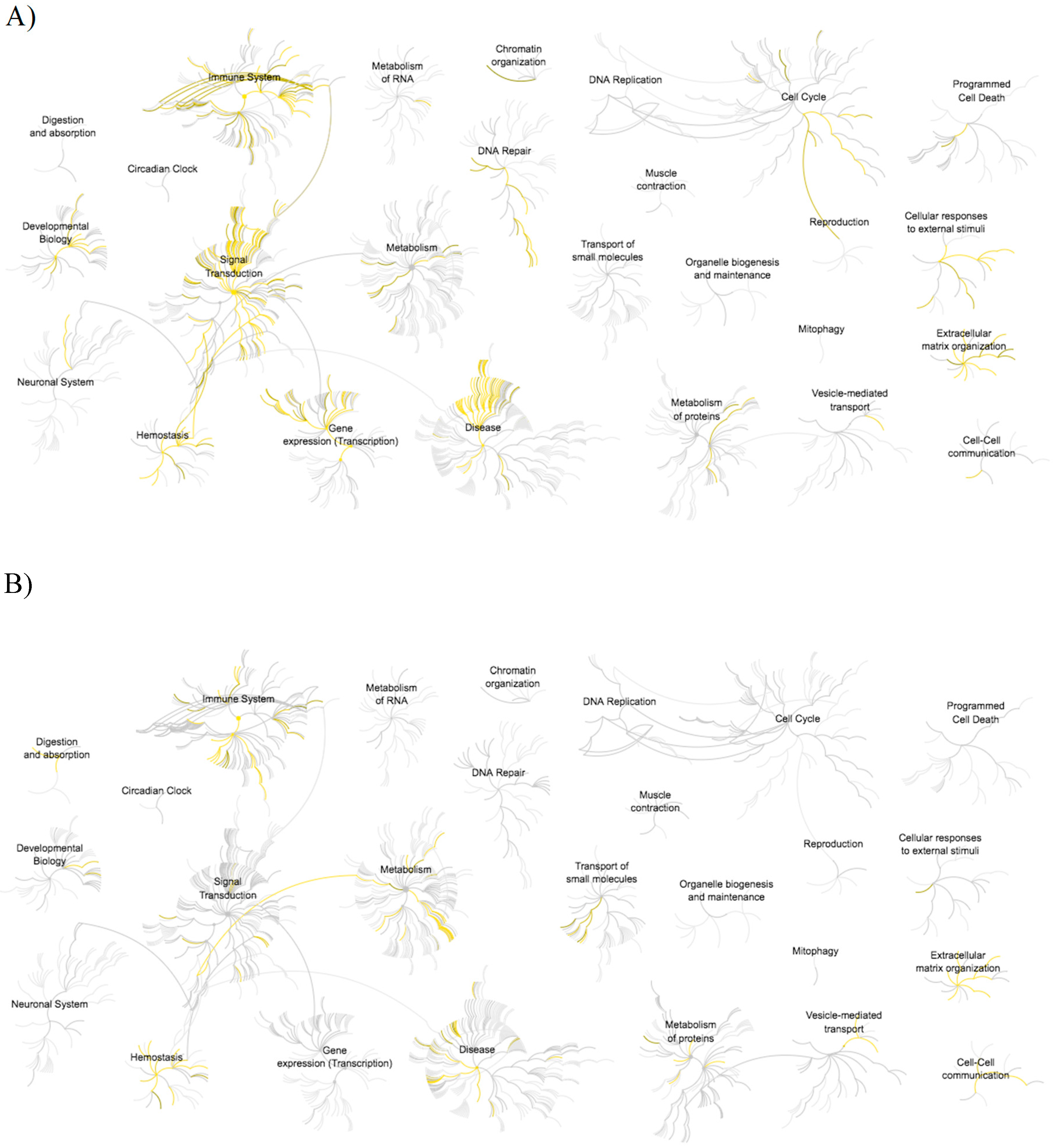
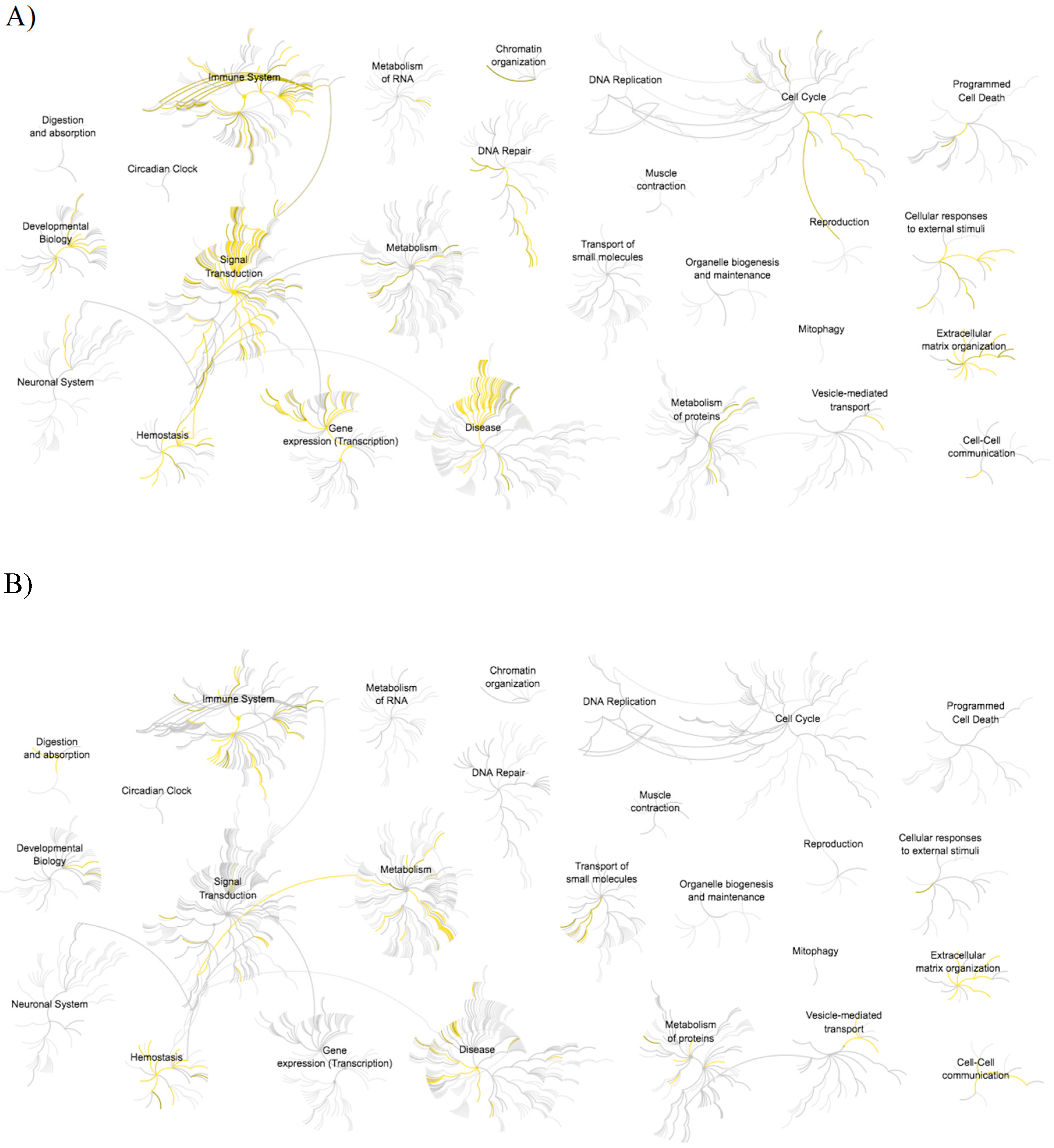
3.3. Pathway Analysis
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thomas, S.; Hao, L.; Ricke, W.A.; Li, L. Biomarker discovery in mass spectrometry-based urinary proteomics. Proteom. Clin. Appl. 2016, 10, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Mann, M. Quantitative analysis of the intra- and inter-individual variability of the normal urinary proteome. J. Proteome Res. 2011, 10, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Legrain, P.; Aebersold, R.; Archakov, A.; Bairoch, A.; Bala, K.; Beretta, L.; Bergeron, J.; Borchers, C.H.; Corthals, G.L.; Costello, C.E.; et al. The human proteome project: Current state and future direction. Mol. Cell. Proteom. 2011, 10, M111-009993. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Allmaier, G.; Apweiler, R.; Attwood, T.; Baumann, M.; Benigni, A.; Bennett, S.E.; Bischoff, R.; Bongcam-Rudloff, E.; Capasso, G.; et al. Recommendations for biomarker identification and qualification in clinical proteomics. Sci. Transl. Med. 2010, 2, 46ps42. [Google Scholar] [CrossRef] [PubMed]
- Deferrari, G.; Repetto, M.; Calvi, C.; Ciabattoni, M.; Rossi, C.; Robaudo, C. Diabetic nephropathy: From micro- to macroalbuminuria. Nephrol. Dial. Transplant. 1998, 13 (Suppl. S8), 11–15. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E. Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N. Engl. J. Med. 1984, 310, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P. Prediction, progression and prevention of diabetic nephropathy. The minkowski lecture 2005. Diabetologia 2006, 49, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (empai) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O'Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Beasley-Green, A. Urine proteomics in the era of mass spectrometry. Int. Neurourol. J. 2016, 20, S70–S75. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Lewis, J.B.; Ravid, M.; Remuzzi, G.; Hunsicker, L.G. for the DEMAND investigators. Prevalence and risk factors for microalbuminuria in a referred cohort of type ii diabetic patients: A global perspective. Kidney Int. 2006, 69, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Mattock, M.B.; Dawnay, A.B.; Kerry, S.; McGuire, A.; Yaqoob, M.; Hitman, G.A.; Hawke, C. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol. Assess. 2005, 9, iii-vi, xiii-163. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Song, X.; Dong, X.; Yang, Y.; Zhang, Z.; Wen, J.; Li, Y.; Zhou, L.; Zhao, N.; Zhu, X.; et al. High prevalence of chronic kidney disease in population-based patients diagnosed with type 2 diabetes in downtown shanghai. J. Diabetes Complicat. 2008, 22, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Bhasin, M.; Dillon, S.T.; Guerrero, M.C.; Nelson, R.G.; Knowler, W.C.; Thadhani, R.; Libermann, T.A. Optimizing a proteomics platform for urine biomarker discovery. Mol. Cell. Proteom. 2010, 9, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [PubMed]
- Tavridou, A.; Georgoulidou, A.; Roumeliotis, A.; Roumeliotis, S.; Giannakopoulou, E.; Papanas, N.; Passadakis, P.; Manolopoulos, V.G.; Vargemezis, V. Association of plasma adiponectin and oxidized low-density lipoprotein with carotid intima-media thickness in diabetic nephropathy. J. Diabetes Res. 2015, 2015, 507265. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gender | n | Age | Alb/Cre | Proteinuria | eGFR | HbA1c |
|---|---|---|---|---|---|---|---|
| DM patient | male | 5 | 68 ± 3 | 5.2 ± 3.1 | - | 2.2 ± 1 | 6.4 ± 0.7 |
| HV | male | 5 | 56 ± 4.4 | ND | - | ND | ND |
| Pathway (Increased) | Count | p Value |
|---|---|---|
| Diseases of signal transduction | 50 | 1.11 × 10−16 |
| Signaling by Receptor Tyrosine Kinase | 53 | 1.11 × 10−16 |
| PI3K/AKT Signaling in Cancer | 25 | 1.11 × 10−16 |
| Signal Transduction | 102 | 4.44 × 10−16 |
| Intracellular signaling by second messengers | 32 | 1.22 × 10−15 |
| Disease | 62 | 1.78 × 10−15 |
| PIP3 activates AKT signaling | 30 | 3.33 × 10−15 |
| Negative regulation of the PI3K/AKT network | 21 | 6.11 × 10−15 |
| Signaling by FGFR in disease | 17 | 4.52 × 10−14 |
| Signaling by SCF-KIT | 14 | 1.62 × 10−13 |
| Pathway (Decreased) | Count | p Value |
| Neutrophil degranulation | 64 | 1.11 × 10−16 |
| Platelet degranulation | 30 | 1.11 × 10−16 |
| Regulation of IGF transport and uptake by IGFBPs | 31 | 1.11 × 10−16 |
| Innate immune system | 100 | 1.11 × 10−16 |
| Response to elevated platelet cytosolic Ca2+ | 30 | 2.22 × 10−16 |
| Post-translational protein phosphorylation | 27 | 4.44 × 10−16 |
| Homeostasis | 63 | 1.67 × 10−12 |
| Platelet activation, signaling and aggregation | 36 | 2.70 × 10−13 |
| Immune system | 121 | 4.97 × 10−10 |
| Binding and Uptake of Ligands by Scavenger Receptor | 21 | 2.21 × 10−9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirao, Y.; Saito, S.; Fujinaka, H.; Miyazaki, S.; Xu, B.; Quadery, A.F.; Elguoshy, A.; Yamamoto, K.; Yamamoto, T. Proteome Profiling of Diabetic Mellitus Patient Urine for Discovery of Biomarkers by Comprehensive MS-Based Proteomics. Proteomes 2018, 6, 9. https://doi.org/10.3390/proteomes6010009
Hirao Y, Saito S, Fujinaka H, Miyazaki S, Xu B, Quadery AF, Elguoshy A, Yamamoto K, Yamamoto T. Proteome Profiling of Diabetic Mellitus Patient Urine for Discovery of Biomarkers by Comprehensive MS-Based Proteomics. Proteomes. 2018; 6(1):9. https://doi.org/10.3390/proteomes6010009
Chicago/Turabian StyleHirao, Yoshitoshi, Suguru Saito, Hidehiko Fujinaka, Shigeru Miyazaki, Bo Xu, Ali F. Quadery, Amr Elguoshy, Keiko Yamamoto, and Tadashi Yamamoto. 2018. "Proteome Profiling of Diabetic Mellitus Patient Urine for Discovery of Biomarkers by Comprehensive MS-Based Proteomics" Proteomes 6, no. 1: 9. https://doi.org/10.3390/proteomes6010009
APA StyleHirao, Y., Saito, S., Fujinaka, H., Miyazaki, S., Xu, B., Quadery, A. F., Elguoshy, A., Yamamoto, K., & Yamamoto, T. (2018). Proteome Profiling of Diabetic Mellitus Patient Urine for Discovery of Biomarkers by Comprehensive MS-Based Proteomics. Proteomes, 6(1), 9. https://doi.org/10.3390/proteomes6010009