
Personalized Proteomics: The Future of Precision Medicine


Abstract
:1. The Age of Precision Medicine
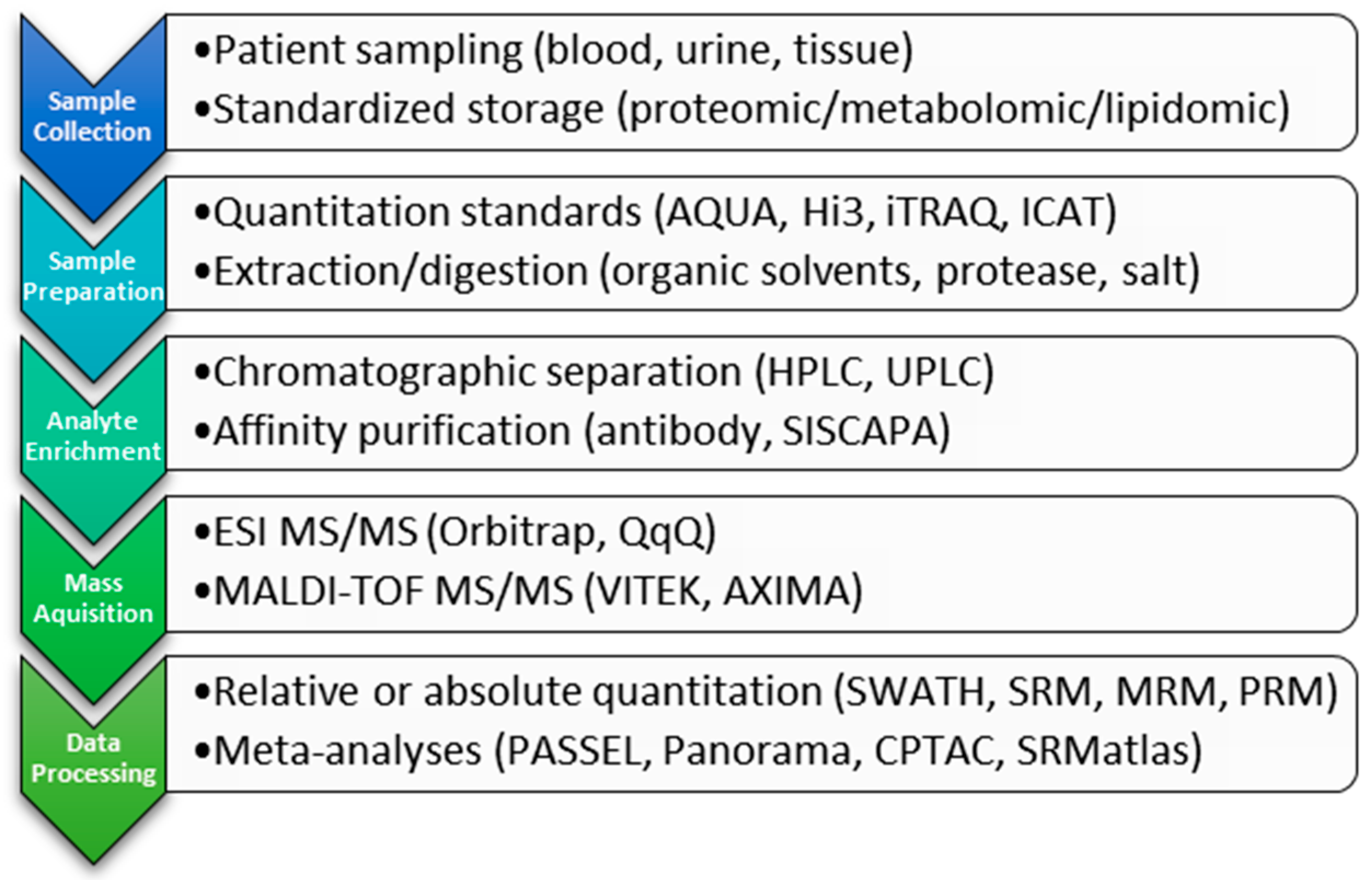
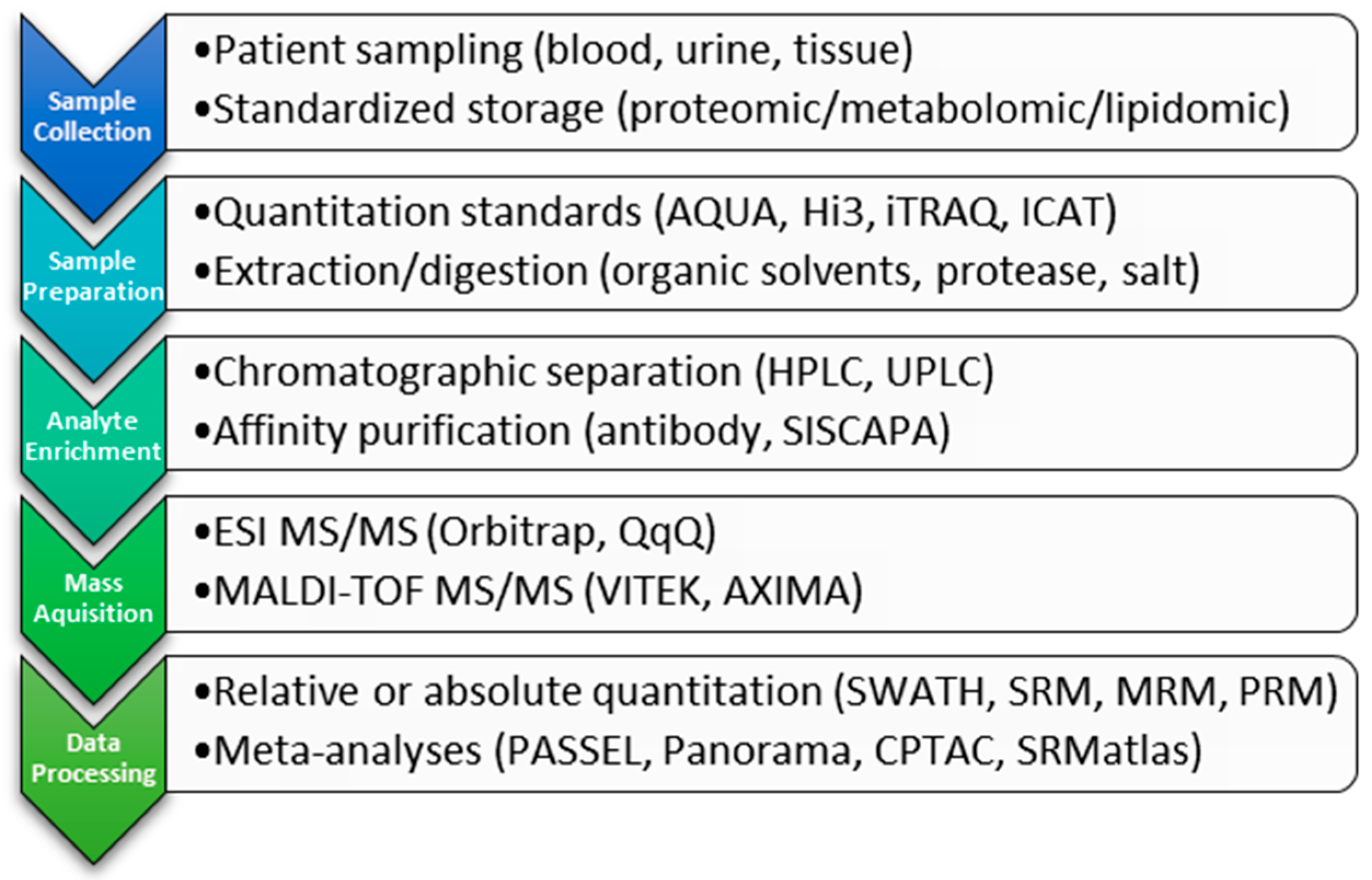
2. Clinical Proteomic Technology
2.1. Ionization
2.2. Detection
2.2.1. Triple Quadrupole
2.2.2. High-Resolution, Mass-Accurate Spectrometers
2.3. Analyte Quantitation Methods
3. Personalized Proteomics
3.1. Proteomics for Biomarker Monitoring
3.2. Proteomics for Diagnosis
3.3. Proteomics to Improve Patient/Therapy Selection
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Specific Genetic Disorders. Available online: https://www.genome.gov/10001204/specific-genetic-disorders/ (accessed on 16 June 2016).
- Novelli, G. Personalized genomic medicine. Intern. Emerg. Med. 2010, 5 (Suppl 1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Personalized medicine. Curr. Opin. Mol. Ther. 2002, 4, 548–558. [Google Scholar] [PubMed]
- Altsuler, D.; Donnelly, P.; Gibbs, R.A.; Yang, H.; Zeng, C.; Shen, Y.; Huang, W.; Wayne, M.M.Y.; Xue, H.; Chee, M.S.; et al. The International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar]
- The Cost of Sequencing a Human Genome. Available online: https://www.genome.gov/27565109/the-cost-of-sequencing-a-human-genome/ (accessed on 16 June 2016).
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Hsi-Yang Fritz, M.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [Green Version]
- 1000 Genomes Project. Available online: http://www.1000genomes.org/data (accessed on 17 June 2016).
- Honey, K. Gina: Making it safe to know what’s in your genes. J. Clin. Investig. 2008, 118, 2369. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Pennisi, E. ENCODE project writes eulogy for junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Julius, M. Precision medicine: Now, not when. Healthc. Manag. Forum 2016, 29, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Herberts, C.A.; van Gaans-van den Brink, J.; van der Heeft, E.; van Wijk, M.; Hoekman, J.; Jaye, A.; Poelen, M.C.M.; Boog, C.J.P.; Roholl, P.J.M.; Whittle, H.; et al. Autoreactivity against induced or upregulated abundant self-peptides in HLA-A*0201 following measles virus infection. Hum. Immunol. 2003, 64, 44–55. [Google Scholar] [CrossRef]
- Wahl, A.; Schafer, F.; Bardet, W.; Hildebrand, W.H. HLA class I molecules reflect an altered host proteome after influenza virus infection. Hum. Immunol. 2010, 71, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Hickman, H.D.; Luis, A.D.; Bardet, W.; Buchli, R.; Battson, C.L.; Shearer, M.H.; Jackson, K.W.; Kennedy, R.C.; Hildebrand, W.H. Cutting edge: Class I presentation of host peptides following HIV infection. J. Immunol. 2003, 171, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Bezbradica, J.S.; Ramos, M.G.; Arico, C.D.; Conant, S.B.; Gilchuk, P.; Gray, J.J.; Zheng, M.; Niu, X.; Hildebrand, W.; et al. Viral infection causes a shift in the self peptide repertoire presented by human MHC class I molecules. Proteom. Clin. Appl. 2015, 9, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Manubhai, K.P.; Kulkarni, V.; Srivastava, S. An overview of innovations and industrial solutions in protein microarray technology. Proteomics 2016, 16, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Brede, C.; Lescuyer, P.; Cocho, J.A.; Vialaret, J.; Bros, P.; Delatour, V.; Hirtz, C. Clinical Mass Spectrometry Proteomics (CMSP) for medical laboratory: What does the future hold? Chim. Acta Int. J. Clin. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lassman, M.E.; McAvoy, T.; Chappell, D.L.; Lee, A.Y.; Zhao, X.X.; Laterza, O.F. The clinical utility of mass spectrometry based protein assays. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 459, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Byrns, S.; Pennington, S.R.; Holmes, D.T.; Anderson, N.L.; Agreste, T.M.; Duffy, M.A. Clinical translation of MS-based, quantitative plasma proteomics: Status, challenges, requirements, and potential. Expert Rev. Proteom. 2016, 13, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, B.; Mindt, S.; Neumaier, M.; Findeisen, P. Clinical applications of MS-based protein quantification. Proteom. Clin. Appl. 2016, 10, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Scherl, A. Clinical protein mass spectrometry. Methods 2015, 81, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Apweiler, R.; Banks, R.E.; Conaway, M.; Coon, J.; Dominiczak, A.; Ehrich, J.H.; Fliser, D.; Girolami, M.; Hermjakob, H.; et al. Clinical proteomics: A need to define the field and to begin to set adequate standards. Proteom. Clin. Appl. 2007, 1, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.; Geromanos, S.J. Absolute quantification of proteins by lcmse: A virtue of parallel MS acquisition. Mol. Cell. Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Torsetnes, S.B.; Levernaes, M.S.; Broughton, M.N.; Paus, E.; Halvorsen, T.G.; Reubsaet, L. Multiplexing determination of small cell lung cancer biomarkers and their isovariants in serum by immunocapture LC-MS/MS. Anal. Chem. 2014, 86, 6983–6992. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Fillmore, T.L.; Gao, Y.; Zhao, R.; He, J.; Schepmoes, A.A.; Nicora, C.D.; Wu, C.; Chambers, J.L.; Moore, R.J.; et al. Long-gradient separations coupled with selected reaction monitoring for highly sensitive, large scale targeted protein quantification in a single analysis. Anal. Chem. 2013, 85, 9196–9203. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Domanski, D.; Percy, A.J.; Yang, J.; Chambers, A.G.; Hill, J.S.; Freue, G.V.; Borchers, C.H. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics 2012, 12, 1222–1243. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.S.; Gerber, S.A.; Gygi, S.P. The absolute quantification strategy: A general procedure for the quantification of proteins and post-translational modifications. Methods 2005, 35, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Jeudy, J.; Salvador, A.; Simon, R.; Jaffuel, A.; Fonbonne, C.; Leonard, J.F.; Gautier, J.C.; Pasquier, O.; Lemoine, J. Overcoming biofluid protein complexity during targeted mass spectrometry detection and quantification of protein biomarkers by mrm cubed (MRM3). Anal. Bioanal. Chem. 2014, 406, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Cox, J.; Mann, M. Proteomics on an orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteom. 2010, 9, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Lange, O.; Denisov, E.; Nolting, D.; Muller, M.; Viner, R.; Schwartz, J.; Remes, P.; Belford, M.; et al. Ultra high resolution linear ion trap orbitrap mass spectrometer (orbitrap elite) facilitates top down LC-MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dillen, L.; Cools, W.; Vereyken, L.; Lorreyne, W.; Huybrechts, T.; de Vries, R.; Ghobarah, H.; Cuyckens, F. Comparison of triple quadrupole and high-resolution TOF-MS for quantification of peptides. Bioanalysis 2012, 4, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Remy-Martin, F.; El Osta, M.; Lucchi, G.; Zeggari, R.; Leblois, T.; Bellon, S.; Ducoroy, P.; Boireau, W. Surface plasmon resonance imaging in arrays coupled with mass spectrometry (Supra-MS): Proof of concept of on-chip characterization of a potential breast cancer marker in human plasma. Anal. Bioanal. Chem. 2012, 404, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, A.; El Osta, M.; Lucchi, G.; Ducoroy, P.; Boireau, W. Immuno-MALDI-MS in human plasma and on-chip biomarker characterizations at the femtomole level. Sensors 2012, 12, 15119–15132. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Kamcheva, E.; Nedelkov, D. Mass spectrometric immunoassay for quantitative determination of protein biomarker isoforms. J. Proteome Res. 2010, 9, 5969–5973. [Google Scholar] [CrossRef] [PubMed]
- Willems, S.M.; van Remoortere, A.; van Zeijl, R.; Deelder, A.M.; McDonnell, L.A.; Hogendoorn, P.C. Imaging mass spectrometry of myxoid sarcomas identifies proteins and lipids specific to tumour type and grade, and reveals biochemical intratumour heterogeneity. J. Pathol. 2010, 222, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Meding, S.; Martin, K.; Gustafsson, O.J.; Eddes, J.S.; Hack, S.; Oehler, M.K.; Hoffmann, P. Tryptic peptide reference data sets for MALDI imaging mass spectrometry on formalin-fixed ovarian cancer tissues. J. Proteome Res. 2013, 12, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Heijs, B.; Abdelmoula, W.M.; Lou, S.; Briaire-de Bruijn, I.H.; Dijkstra, J.; Bovee, J.V.; McDonnell, L.A. Histology-guided high-resolution matrix-assisted laser desorption ionization mass spectrometry imaging. Anal. Chem. 2015, 87, 11978–11983. [Google Scholar] [CrossRef] [PubMed]
- Makarov, A. Electrostatic axially harmonic orbital trapping: A high-performance technique of mass analysis. Anal. Chem. 2000, 72, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Domon, B. Advances in high-resolution quantitative proteomics: Implications for clinical applications. Expert Rev. Proteom. 2015, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, J.A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.; Wechalekar, A.; Whelan, C.; Hawkins, P.N.; Dogan, A.; Gillmore, J.D. A comparison of immunohistochemistry and mass spectrometry for determining the amyloid fibril protein from formalin-fixed biopsy tissue. J. Clin. Pathol. 2015, 68, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Rush, J.; Stemman, O.; Kirschner, M.W.; Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 2003, 100, 6940–6945. [Google Scholar] [CrossRef] [PubMed]
- Muntel, J.; Fromion, V.; Goelzer, A.; Maabeta, S.; Mader, U.; Buttner, K.; Hecker, M.; Becher, D. Comprehensive absolute quantification of the cytosolic proteome of bacillus subtilis by data independent, parallel fragmentation in liquid chromatography/mass spectrometry (LC/MS(e)). Mol. Cell. Proteom. 2014, 13, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Kretschy, D.; Koellensperger, G.; Hann, S. Elemental labelling combined with liquid chromatography inductively coupled plasma mass spectrometry for quantification of biomolecules: A review. Anal. Chim. Acta 2012, 750, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Eckels, J.; Taylor, G.K.; Shulman, N.J.; Stergachis, A.B.; Joyner, S.A.; Yan, P.; Whiteaker, J.R.; Halusa, G.N.; Schilling, B.; et al. Panorama: A targeted proteomics knowledge base. J. Proteome Res. 2014, 13, 4205–4210. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, J.R.; Halusa, G.N.; Hoofnagle, A.N.; Sharma, V.; MacLean, B.; Yan, P.; Wrobel, J.A.; Kennedy, J.; Mani, D.R.; Zimmerman, L.J.; et al. CPTAC assay portal: A repository of targeted proteomic assays. Nat. Methods 2014, 11, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human SRMAtlas: A resource of targeted assays to quantify the complete human proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.; Deutsch, E.W.; Kreisberg, R.; Sun, Z.; Campbell, D.S.; Mendoza, L.; Kusebauch, U.; Brusniak, M.-Y.; Hüttenhain, R.; Schiess, R.; et al. Passel: The peptideatlas SRMExperiment library. Proteomics 2012, 12, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G.; Haines, L.R.; Hardie, D.B.; Olafson, R.W.; Pearson, T.W. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 2004, 3, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, R.M.; Zhao, L.; Ivey, R.G.; Voytovich, U.J.; Kennedy, J.; Yan, P.; Lin, C.; Whiteaker, J.R.; Paulovich, A.G. Commercially available antibodies can be applied in quantitative multiplexed peptide immunoaffinity enrichment targeted mass spectrometry assays. Proteomics 2016. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Moghaddas Gholami, A.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, C. The potential clinical impact of the tissue-based map of the human proteome. Expert Rev. Proteom. 2015, 12, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Rezai, T.; Krastins, B.; Sarracino, D.; Athanas, M.; Russo, P.; Zhang, H.; Tian, Y.; Li, Y.; Kulasingam, V.; et al. Interlaboratory reproducibility of selective reaction monitoring assays using multiple upfront analyte enrichment strategies. J. Proteome Res. 2012, 11, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Rezai, T.; Krastins, B.; Sarracino, D.; Athanas, M.; Russo, P.; Ross, M.M.; Zhang, H.; Tian, Y.; Kulasingam, V.; et al. Platform for establishing interlaboratory reproducibility of selected reaction monitoring-based mass spectrometry peptide assays. J. Proteome Res. 2010, 9, 6678–6688. [Google Scholar] [CrossRef] [PubMed]
- Abbatiello, S.E.; Schilling, B.; Mani, D.R.; Zimmerman, L.J.; Hall, S.C.; MacLean, B.; Albertolle, M.; Allen, S.; Burgess, M.; Cusack, M.P.; et al. Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol. Cell. Proteom. 2015, 14, 2357–2374. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, A.N.; Becker, J.O.; Oda, M.N.; Cavigiolio, G.; Mayer, P.; Vaisar, T. Multiple-reaction monitoring–mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin. Chem. 2012, 58, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Sun, X.; Gao, Y.; Fillmore, T.L.; Schepmoes, A.A.; Zhao, R.; He, J.; Moore, R.J.; Kagan, J.; Rodland, K.D.; et al. Targeted quantification of low ng/mL level proteins in human serum without immunoaffinity depletion. J. Proteome Res. 2013, 12, 3353–3361. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Su, D.; Liu, T.; Tang, K.; Camp, D.G.; Qian, W.-J.; Smith, R.D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 12, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M. LC-MS/MS for protein and peptide quantification in clinical chemistry. J. Chromatogr. B 2012, 883–884, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D. Mass spectrometry-based protein assays for in vitro diagnostic testing. Expert Rev. Mol. Diagn. 2012, 12, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Boja, E.S.; Rodriguez, H. Mass spectrometry-based targeted quantitative proteomics: Achieving sensitive and reproducible detection of proteins. Proteomics 2012, 12, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Schoenhoff, F.S.; Savage, W.J.; Zhang, P.; Van Eyk, J.E. Multiplex assays for biomarker research and clinical application: Translational science coming of age. Proteom. Clin. Appl. 2010, 4, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Krastins, B.; Prakash, A.; Sarracino, D.A.; Nedelkov, D.; Niederkofler, E.E.; Kiernan, U.A.; Nelson, R.; Vogelsang, M.S.; Vadali, G.; Garces, A.; et al. Rapid development of sensitive, high-throughput, quantitative and highly selective mass spectrometric targeted immunoassays for clinically important proteins in human plasma and serum. Clin. Biochem. 2013, 46, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.B.; French, D. Implementation of liquid chromatography/mass spectrometry into the clinical laboratory. Clin. Chim. Acta 2013, 420, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Van Den Ouweland, J.M.W.; Kema, I.P. The role of liquid chromatography–tandem mass spectrometry in the clinical laboratory. J. Chromatogr. B 2012, 883–884, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Strathmann, F.G.; Hoofnagle, A.N. Current and future applications of mass spectrometry to the clinical laboratory. Am. J. Clin. Pathol. 2011, 136, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Seger, C. Usage and limitations of liquid chromatography-tandem mass spectrometry (LC-MS/MS) in clinical routine laboratories. Wien. Med. Wochenschr. 2012, 162, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.O.; Hoofnagle, A.N. Replacing immunoassays with tryptic digestion-peptide immunoaffinity enrichment and LC-MS/MS. Bioanalysis 2012, 4, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, A.N.; Wener, M.H. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J. Immunol. Methods 2009, 347, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Schaab, M.R.; Nelson, R.W.; Nedelkov, D. Development of multiplex mass spectrometric immunoassay for detection and quantification of apolipoproteins C-I, C-II, C-III and their proteoforms. Methods 2015, 81, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Vilà-Rico, M.; Colomé-Calls, N.; Martín-Castel, L.; Gay, M.; Azorín, S.; Vilaseca, M.; Planas, A.; Canals, F. Quantitative analysis of post-translational modifications in human serum transthyretin associated with familial amyloidotic polyneuropathy by targeted LC-MS and intact protein MS. J. Proteom. 2015, 127, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.H.; Wu, J.J.; Ren, H.M.; Wang, J.; Ding, Z.T.; Jiang, Y.P. Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp. Ther. Med. 2016, 11, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Wasinger, V.C.; Leong, R.W. Current application of proteomics in biomarker discovery for inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2016, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bandu, R.; Mok, H.J.; Kim, K.P. Phospholipids as cancer biomarkers: Mass spectrometry-based analysis. Mass Spectrom. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, A.; Pasic, M.D.; Yousef, G.M. Proteomics and peptidomics: Moving toward precision medicine in urological malignancies. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hanash, S. Mass spectrometry based proteomics for absolute quantification of proteins from tumor cells. Methods 2015, 81, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Friese, R.S.; Rao, F.; Khandrika, S.; Thomas, B.; Ziegler, M.G.; Schmid-Schonbein, G.W.; O’Connor, D.T. Matrix metalloproteinases: Discrete elevations in essential hypertension and hypertensive end-stage renal disease. Clin. Exp. Hypertens. 2009, 31, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Hobeika, M.J.; Thompson, R.W.; Muhs, B.E.; Brooks, P.C.; Gagne, P.J. Matrix metalloproteinases in peripheral vascular disease. J. Vasc. Surg. 2007, 45, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.B.; Barrett-Connor, E.; Clopton, P.; Laughlin, G.A.; Ix, J.H.; Maisel, A.S. Plasma neutrophil gelatinase-associated lipocalin is independently associated with cardiovascular disease and mortality in community-dwelling older adults: The rancho bernardo study. J. Am. Coll. Cardiol. 2012, 59, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- De Franciscis, S.; Serra, R. Matrix metalloproteinases and endothelial dysfunction: The search for new prognostic markers and for new therapeutic targets for vascular wall imbalance. Thromb. Res. 2015, 136, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, P.R.; Verma, M.; Zhao, Y.; Srivastava, S. Proteomics for cancer biomarker discovery. Clin. Chem. 2002, 48, 1160–1169. [Google Scholar] [PubMed]
- Longoria, T.C.; Ueland, F.R.; Zhang, Z.; Chan, D.W.; Smith, A.; Fung, E.T.; Munroe, D.G.; Bristow, R.E. Clinical performance of a multivariate index assay for detecting early-stage ovarian cancer. Am. J. Obstet. Gynecol. 2014, 210, 78.e1–78.e9. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Cernei, N.; Heger, Z.; Gumulec, J.; Zitka, O.; Masarik, M.; Babula, P.; Eckschlager, T.; Stiborova, M.; Kizek, R.; Adam, V. Sarcosine as a potential prostate cancer biomarker—A review. Int. J. Mol. Sci. 2013, 14, 13893–13908. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Kippen, A.D.; Cerini, F.; Vadas, L.; Stöcklin, R.; Vu, L.; Offord, R.E.; Rose, K. Development of an isotope dilution assay for precise determination of insulin, c-peptide, and proinsulin levels in non-diabetic and type ii diabetic individuals with comparison to immunoassay. J. Biol. Chem. 1997, 272, 12513–12522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., III. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, M.M.; Rockwood, A.L.; Strathmann, F.G.; Frank, E.L.; Straseski, J.A.; Meikle, A.W. LC-MS/MS measurement of parathyroid hormone–related peptide. Clin. Chem. 2016, 62, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Wieringa, G.E.; Sturgeon, C.M.; Trainer, P.J. The harmonisation of growth hormone measurements: Taking the next steps. Clin. Chim. Acta 2014, 432, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Arsene, C.G.; Kratzsch, J.; Henrion, A. Mass spectrometry—An alternative in growth hormone measurement. Bioanalysis 2014, 6, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.G.; Bruns, D.E.; Hortin, G.L.; Sandberg, S.; Aakre, K.M.; McQueen, M.J.; Itoh, Y.; Lieske, J.C.; Seccombe, D.W.; Jones, G.; et al. Current issues in measurement and reporting of urinary albumin excretion. Clin. Chem. 2009, 55, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.R.; Barnidge, D.R.; Murray, D.L. Detecting monoclonal immunoglobulins in human serum using mass spectrometry. Methods 2015, 81, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Stringer, K.A.; McKay, R.T.; Karnovsky, A.; Quemerais, B.; Lacy, P. Metabolomics and its application to acute lung diseases. Front. Immunol. 2016, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Chui, H.; Domish, L.; Hernandez, D.; Wang, G. Recent development of mass spectrometry and proteomics applications in identification and typing of bacteria. Proteom. Clin. Appl. 2016, 10, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Alispahic, M.; Christensen, H.; Bisgaard, M.; Hess, M.; Hess, C. MALDI-TOF mass spectrometry confirms difficulties in separating species of the avibacterium genus. Avian Pathol. 2014, 43, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Samb-Ba, B.; Mazenot, C.; Gassama-Sow, A.; Dubourg, G.; Richet, H.; Hugon, P.; Lagier, J.C.; Raoult, D.; Fenollar, F. MALDI-TOF identification of the human gut microbiome in people with and without diarrhea in senegal. PLoS ONE 2014, 9, e87419. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhang, H.; He, L.; Peng, X.; Wang, Y.; Xue, G.; Su, P.; Zhang, J. High natural variability bacteria identification and typing: Helicobacter pylori analysis based on peptide mass fingerprinting. J. Proteom. 2014, 98, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Kooken, J.; Fox, K.; Fox, A.; Altomare, D.; Creek, K.; Wunschel, D.; Pajares-Merino, S.; Martinez-Ballesteros, I.; Garaizar, J.; Oyarzabal, O.; et al. Identification of staphylococcal species based on variations in protein sequences (mass spectrometry) and DNA sequence (soda microarray). Mol. Cell. Probes 2014, 28, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lasch, P.; Fleige, C.; Stammler, M.; Layer, F.; Nubel, U.; Witte, W.; Werner, G. Insufficient discriminatory power of MALDI-TOF mass spectrometry for typing of enterococcus faecium and staphylococcus aureus isolates. J. Microbiol. Methods 2014, 100, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Lima Ede, O.; de Macedo, C.S.; Esteves, C.Z.; de Oliveira, D.N.; Pessolani, M.C.; Nery, J.A.; Sarno, E.N.; Catharino, R.R. Skin imprinting in silica plates: A potential diagnostic methodology for leprosy using high-resolution mass spectrometry. Anal. Chem. 2015, 87, 3585–3592. [Google Scholar] [CrossRef] [PubMed]
- Barbuddhe, S.B.; Maier, T.; Schwarz, G.; Kostrzewa, M.; Hof, H.; Domann, E.; Chakraborty, T.; Hain, T. Rapid identification and typing of listeria species by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 2008, 74, 5402–5407. [Google Scholar] [CrossRef] [PubMed]
- Moura, H.; Terilli, R.R.; Woolfitt, A.R.; Williamson, Y.M.; Wagner, G.; Blake, T.A.; Solano, M.I.; Barr, J.R. Proteomic analysis and label-free quantification of the large clostridium difficile toxins. Int. J. Proteom. 2013, 293782. [Google Scholar] [CrossRef]
- Wang, D.; Krilich, J.; Baudys, J.; Barr, J.R.; Kalb, S.R. Enhanced detection of type C botulinum neurotoxin by the Endopep-MS assay through optimization of peptide substrates. Bioorg. Med. Chem. 2015, 23, 3667–3673. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Baudys, J.; Wang, D.; Barr, J.R. Recommended mass spectrometry-based strategies to identify botulinum neurotoxin-containing samples. Toxins 2015, 7, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Herrenthey, A.; Maier, T.; Gessler, F.; Schaumann, R.; Bohnel, H.; Kostrzewa, M.; Kruger, M. Challenging the problem of clostridial identification with matrix-assisted laser desorption and ionization-time-of-flight mass spectrometry (MALDI-TOF MS). Anaerobe 2008, 14, 242–249. [Google Scholar] [CrossRef] [PubMed]
- McFarland, M.A.; Andrzejewski, D.; Musser, S.M.; Callahan, J.H. Platform for identification of salmonella serovar differentiating bacterial proteins by top-down mass spectrometry: S. Typhimurium vs S. Heidelberg. Anal. Chem. 2014, 86, 6879–6886. [Google Scholar] [CrossRef] [PubMed]
- Fagerquist, C.K.; Zaragoza, W.J.; Sultan, O.; Woo, N.; Quinones, B.; Cooley, M.B.; Mandrell, R.E. Top-down proteomic identification of Shiga toxin 2 subtypes from Shiga toxin-producing Escherichia coli by matrix-assisted laser desorption ionization-tandem time of flight mass spectrometry. Appl. Environ. Microbiol. 2014, 80, 2928–2940. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Sloan, A.; Peterson, L.; McCorrister, S.; Robinson, A.; Walker, M.; Drew, T.; McCrea, J.; Chui, L.; Wylie, J.; et al. Comparative study of traditional flagellum serotyping and liquid chromatography-tandem mass spectrometry-based flagellum typing with clinical Escherichia coli isolates. J. Clin. Microbiol. 2014, 52, 2275–2278. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.G.; Kruczkiewicz, P.; Guan, C.; McCorrister, S.J.; Chong, P.; Wylie, J.; van Caeseele, P.; Tabor, H.A.; Snarr, P.; Gilmour, M.W.; et al. Evaluation of MALDI-TOF mass spectroscopy methods for determination of Escherichia coli pathotypes. J. Microbiol. Methods 2013, 94, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Kooken, J.; Fox, K.; Fox, A.; Wunschel, D. Assessment of marker proteins identified in whole cell extracts for bacterial speciation using liquid chromatography electrospray ionization tandem mass spectrometry. Mol. Cell. Probes 2014, 28, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Popp, C.; Sparbier, K.; Lange, C.; Kostrzewa, M.; Schubert, S. Evaluation of matrix-assisted laser desorption ionization-time of flight mass spectrometry for rapid detection of beta-lactam resistance in Enterobacteriaceae derived from blood cultures. J. Clin. Microbiol. 2014, 52, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Gekenidis, M.T.; Studer, P.; Wuthrich, S.; Brunisholz, R.; Drissner, D. Beyond the matrix-assisted laser desorption ionization (MALDI) biotyping workflow: In search of microorganism-specific tryptic peptides enabling discrimination of subspecies. Appl. Environ. Microbiol. 2014, 80, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Chui, H.; Chan, M.; Hernandez, D.; Chong, P.; McCorrister, S.; Robinson, A.; Walker, M.; Peterson, L.A.; Ratnam, S.; Haldane, D.J.; et al. Rapid, Sensitive, and Specific Escherichia coli H Antigen Typing by Matrix-Assisted Laser Desorption Ionization-Time of Flight-Based Peptide Mass Fingerprinting. J. Clin. Microbiol. 2015, 53, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Conway, G.C.; Smole, S.C.; Sarracino, D.A.; Arbeit, R.D.; Leopold, P.E. Phyloproteomics: Species identification of Enterobacteriaceae using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Mol. Microbiol. Biotechnol. 2001, 3, 103–112. [Google Scholar] [PubMed]
- Richter, S.S.; Sercia, L.; Branda, J.A.; Burnham, C.A.; Bythrow, M.; Ferraro, M.J.; Garner, O.B.; Ginocchio, C.C.; Jennemann, R.; Lewinski, M.A.; et al. Identification of Enterobacteriaceae by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using the VITEK MS system. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Zautner, A.E.; Masanta, W.O.; Tareen, A.M.; Weig, M.; Lugert, R.; Gross, U.; Bader, O. Discrimination of multilocus sequence typing-based Campylobacter jejuni subgroups by MALDI-TOF mass spectrometry. BMC Microbiol. 2013, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Segawa, S.; Sawai, S.; Murata, S.; Nishimura, M.; Beppu, M.; Sogawa, K.; Watanabe, M.; Satoh, M.; Matsutani, T.; Kobayashi, M.; et al. Direct application of MALDI-TOF mass spectrometry to cerebrospinal fluid for rapid pathogen identification in a patient with bacterial meningitis. Clin. Chim. Acta Int. J. Clin. Chem. 2014, 435, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Nyvang Hartmeyer, G.; Kvistholm Jensen, A.; Bocher, S.; Damkjaer Bartels, M.; Pedersen, M.; Engell Clausen, M.; Abdul-Redha, R.; Dargis, R.; Schouenborg, P.; Hojlyng, N.; et al. Mass spectrometry: Pneumococcal meningitis verified and Brucella species identified in less than half an hour. Scand. J. Infect. Dis. 2010, 42, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, S.; Dicuonzo, G.; D’Agostino, A.; Avola, A.; Crea, F.; Palazzo, C.; Dedej, E.; De Florio, L. Turnaround time of positive blood cultures after the introduction of matrix-assisted laser desorption-ionization time-of-flight mass spectrometry. New Microbiol. 2015, 38, 379–386. [Google Scholar] [PubMed]
- DeMarco, M.L.; Ford, B.A. Beyond identification: Emerging and future uses for MALDI-TOF mass spectrometry in the clinical microbiology laboratory. Clin. Lab. Med. 2013, 33, 611–628. [Google Scholar] [CrossRef] [PubMed]
- March, G.A.; Garcia-Loygorri, M.C.; Simarro, M.; Gutierrez, M.P.; Orduna, A.; Bratos, M.A. A new approach to determine the susceptibility of bacteria to antibiotics directly from positive blood culture bottles in two hours. J. Microbiol. Methods 2015, 109, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.C.; Williams, G.J.; Jones, M.; Codarini, M.; Macaskill, P.; Hayen, A.; Irwig, L.; Fitzgerald, D.A.; Isaacs, D.; McCaskill, M. The accuracy of clinical symptoms and signs for the diagnosis of serious bacterial infection in young febrile children: Prospective cohort study of 15,781 febrile illnesses. BMJ 2010, 340, c1594. [Google Scholar] [CrossRef] [PubMed]
- Van den Bruel, A.; Thompson, M.J.; Haj-Hassan, T.; Stevens, R.; Moll, H.; Lakhanpaul, M.; Mant, D. Diagnostic value of laboratory tests in identifying serious infections in febrile children: Systematic review. BMJ 2011, 342, d3082. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.; Wertheim, H.F.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Oved, K.; Cohen, A.; Boico, O.; Navon, R.; Friedman, T.; Etshtein, L.; Kriger, O.; Bamberger, E.; Fonar, Y.; Yacobov, R.; et al. A novel host-proteome signature for distinguishing between acute bacterial and viral infections. PLoS ONE 2015, 10, e0120012. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Sanchez, F.; Valenzuela-Mendez, B.; Rodriguez-Gutierrez, J.F.; Rello, J. Personalized medicine in severe influenza. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, P.; Hausfater, P.; Amin, D.; Amin, A.; Haubitz, S.; Faessler, L.; Kutz, A.; Conca, A.; Reutlinger, B.; Canavaggio, P.; et al. Biomarkers from distinct biological pathways improve early risk stratification in medical emergency patients: The multinational, prospective, observational TRIAGE study. Crit. Care 2015, 19, 377. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. New Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Ezan, E.; Dubois, M.; Becher, F. Bioanalysis of recombinant proteins and antibodies by mass spectrometry. Analyst 2009, 134, 825–834. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Zhang, M.; Qu, J. Toward Sensitive and Accurate Analysis of Antibody Biotherapeutics by Liquid Chromatography Coupled with Mass Spectrometry. Drug Metabol. Dispos. 2014, 42, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Mehl, J.; Zhu, Y.; Xin, B.; Olah, T. Application and challenges in using LC-MS assays for absolute quantitative analysis of therapeutic proteins in drug discovery. Bioanalysis 2014, 6, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Van Den Broek, I.; Niessen, W.M.A.; van Dongen, W.D. Bioanalytical LC-MS/MS of protein-based biopharmaceuticals. J. Chromatogr. B 2013, 929, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Lassman, M.E.; McAvoy, T.; Lee, A.Y.; Chappell, D.; Wong, O.; Zhou, H.; Reyes-Soffer, G.; Ginsberg, H.N.; Millar, J.S.; Rader, D.J.; et al. Practical immunoaffinity-enrichment LC-MS for measuring protein kinetics of low-abundance proteins. Clin. Chem. 2014, 60, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.S.; Reyes-Soffer, G.; Jumes, P.; Dunbar, R.L.; deGoma, E.M.; Baer, A.L.; Karmally, W.; Donovan, D.S.; Rafeek, H.; Pollan, L.; et al. Anacetrapib lowers LDL by increasing ApoB clearance in mildly hypercholesterolemic subjects. J. Clin. Investig. 2015, 125, 2510–2522. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Castro-Perez, J.; Lassman, M.E.; Thomas, T.; Li, W.; McLaughlin, T.; Dan, X.; Jumes, P.; Wagner, J.A.; Gutstein, D.E.; et al. Measurement of apo(a) kinetics in human subjects using a microfluidic device with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Gratwohl, A.; Dohler, B.; Stern, M.; Opelz, G. H-Y as a minor histocompatibility antigen in kidney transplantation: A retrospective cohort study. Lancet 2008, 372, 49–53. [Google Scholar] [CrossRef]
- Dierselhuis, M.; Goulmy, E. The relevance of minor histocompatibility antigens in solid organ transplantation. Curr. Opin. Organ Transplant. 2009, 14, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Gilchuk, P.; Dragovic, S.M.; Joyce, S. Minor histocompatibility antigens: Presentation principles, recognition logic and the potential for a healing hand. Curr. Opin. Organ Transplant. 2010, 15, 512–525. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Category | NGS/Genetics a | ELISA b | Flow Cytometry c | Microarray d | MS e |
|---|---|---|---|---|---|
| Protein mutations | Only if encoded genetically | Sequence or structure recognized by antibody | Sequence or structure recognized by antibody & cellular | Only in sequence | Only in sequence |
| PTMs | Binding site may be identifiable | If differentially recognized by antibody | If differentially recognized by antibody & cellular | Reported for select PTMs | YES |
| Expression level | Inferred based on promoter/enhancers | YES | YES | YES | YES |
| Metabolites | Predicted | If unique antibody is available | If unique antibody is available & cellular | Selected | YES |
| Metabolic flux | Predicted | YES | Only intracellular | YES | YES |
| Enzymatic activity | Predicted | YES | No | Potential | YES |
| Number of analytes/test | Millions of bases | <100 | 15/40f | Thousands | Thousands |
| Targeted biomarkers | YES | YES | YES | YES | YES |
| Discovery analysis | YES | No | No | Limited | YES |
| Differentiate heterogeneous mixture | No | If differentially recognized by antibody | If differentially recognized by antibody & cellular | YES | YES |
| Serial analysis of patient samples | No new information is obtained | YES | YES | YES | YES |
| Pharmacokinetics | No | YES | No | Potential | YES |
| Therapeutic monitoring | No | YES | If cellular | Potential | YES |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, T.T.; Spencer, C.T. Personalized Proteomics: The Future of Precision Medicine. Proteomes 2016, 4, 29. https://doi.org/10.3390/proteomes4040029
Duarte TT, Spencer CT. Personalized Proteomics: The Future of Precision Medicine. Proteomes. 2016; 4(4):29. https://doi.org/10.3390/proteomes4040029
Chicago/Turabian StyleDuarte, Trevor T., and Charles T. Spencer. 2016. "Personalized Proteomics: The Future of Precision Medicine" Proteomes 4, no. 4: 29. https://doi.org/10.3390/proteomes4040029
APA StyleDuarte, T. T., & Spencer, C. T. (2016). Personalized Proteomics: The Future of Precision Medicine. Proteomes, 4(4), 29. https://doi.org/10.3390/proteomes4040029