
Activity-Based Proteomics Reveals Heterogeneous Kinome and ATP-Binding Proteome Responses to MEK Inhibition in KRAS Mutant Lung Cancer

 , , and
, , and
Abstract
:1. Introduction
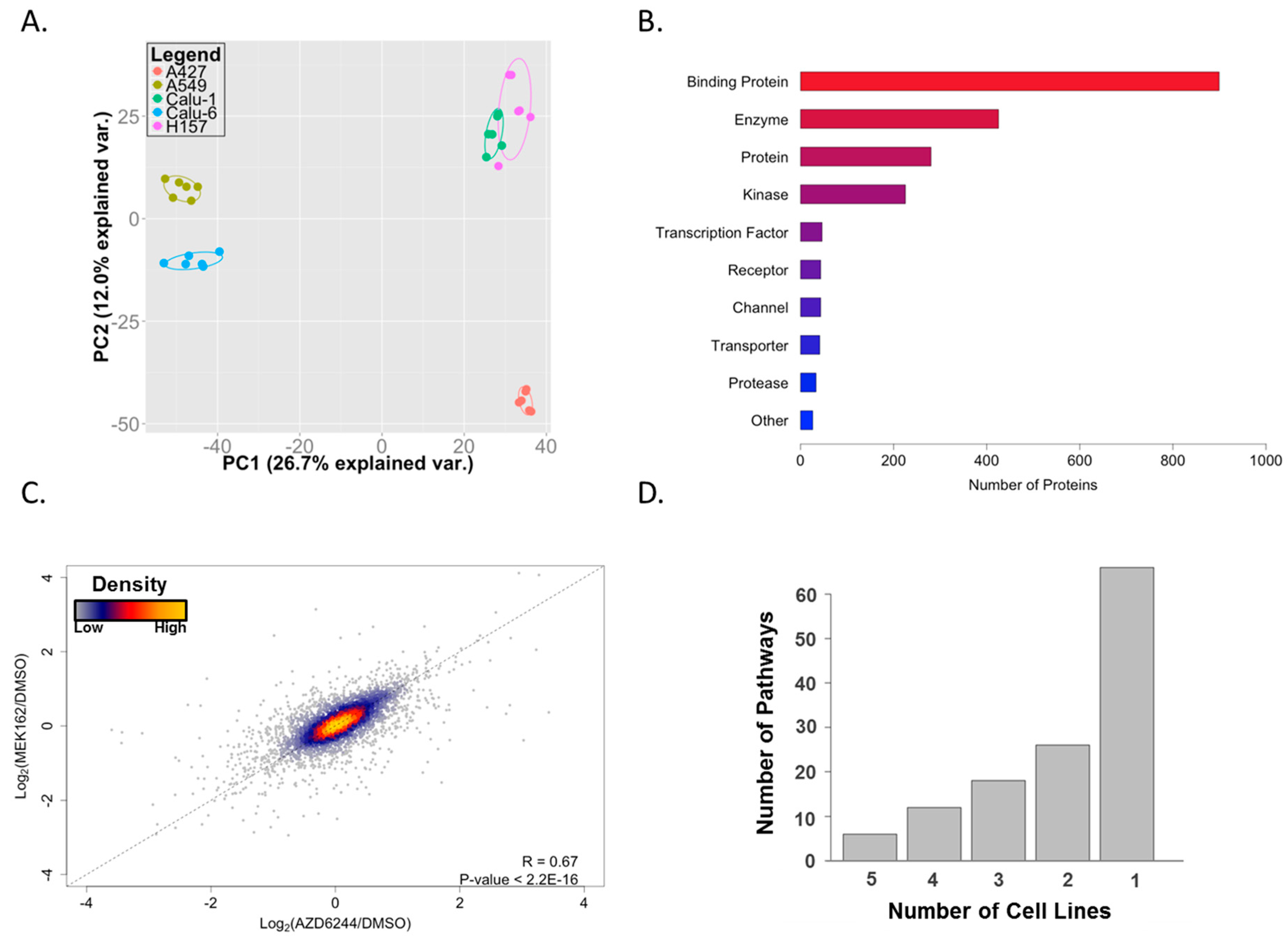
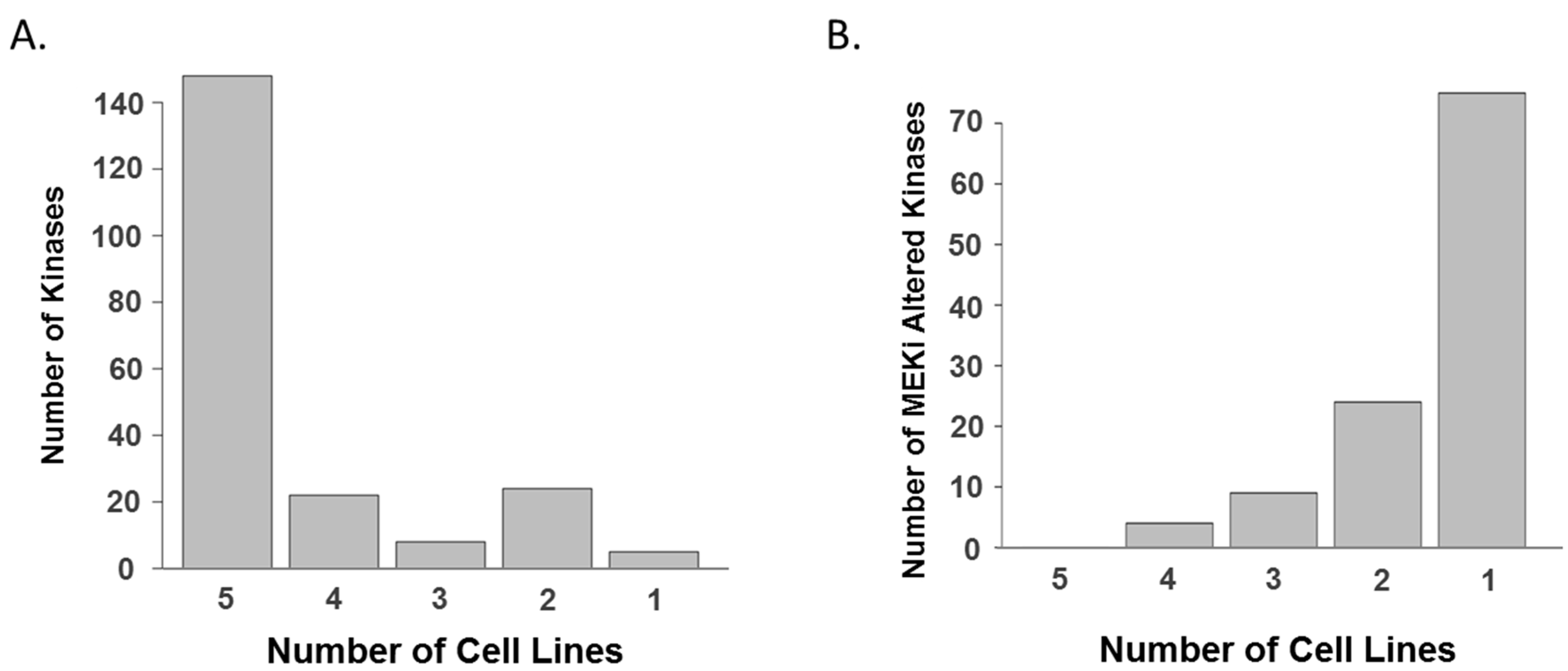
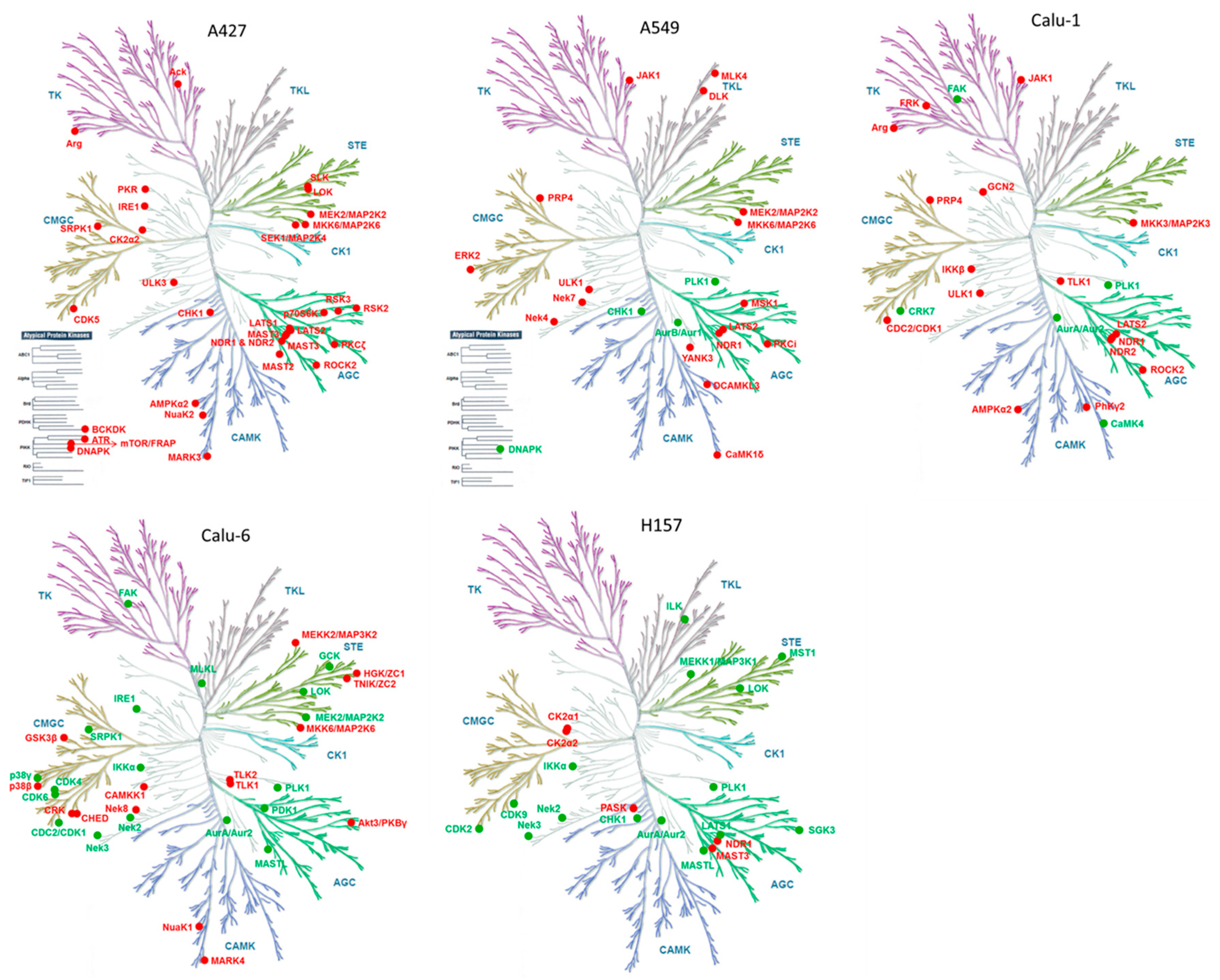
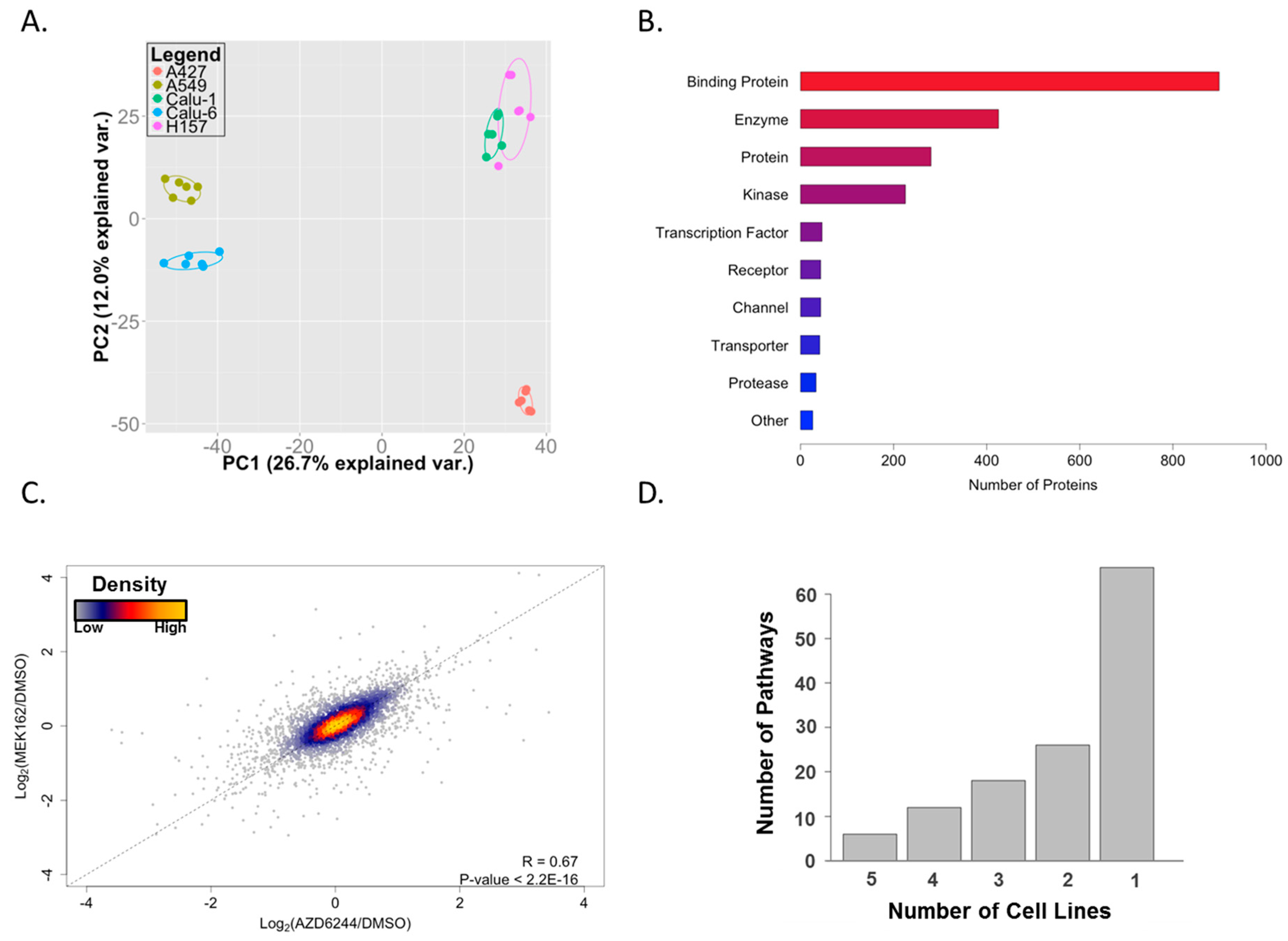
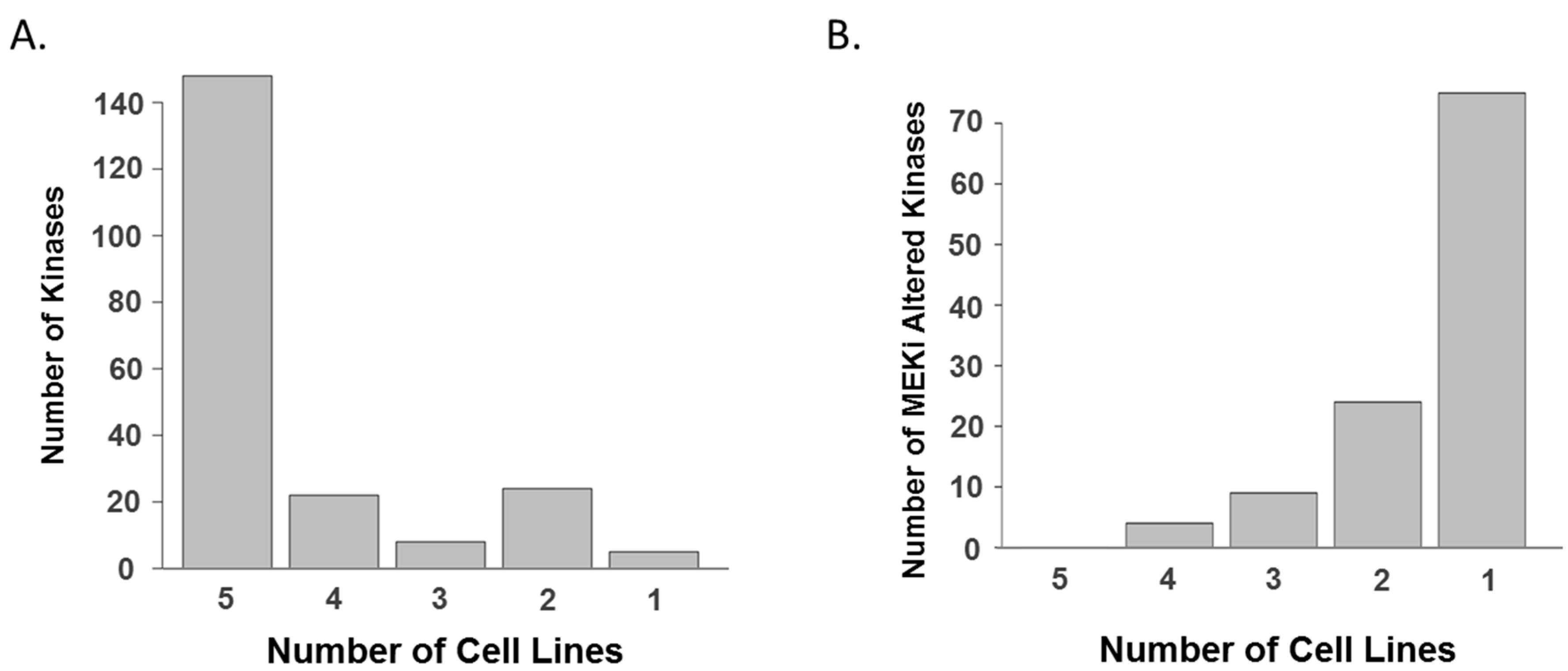
2. Results and Discussion
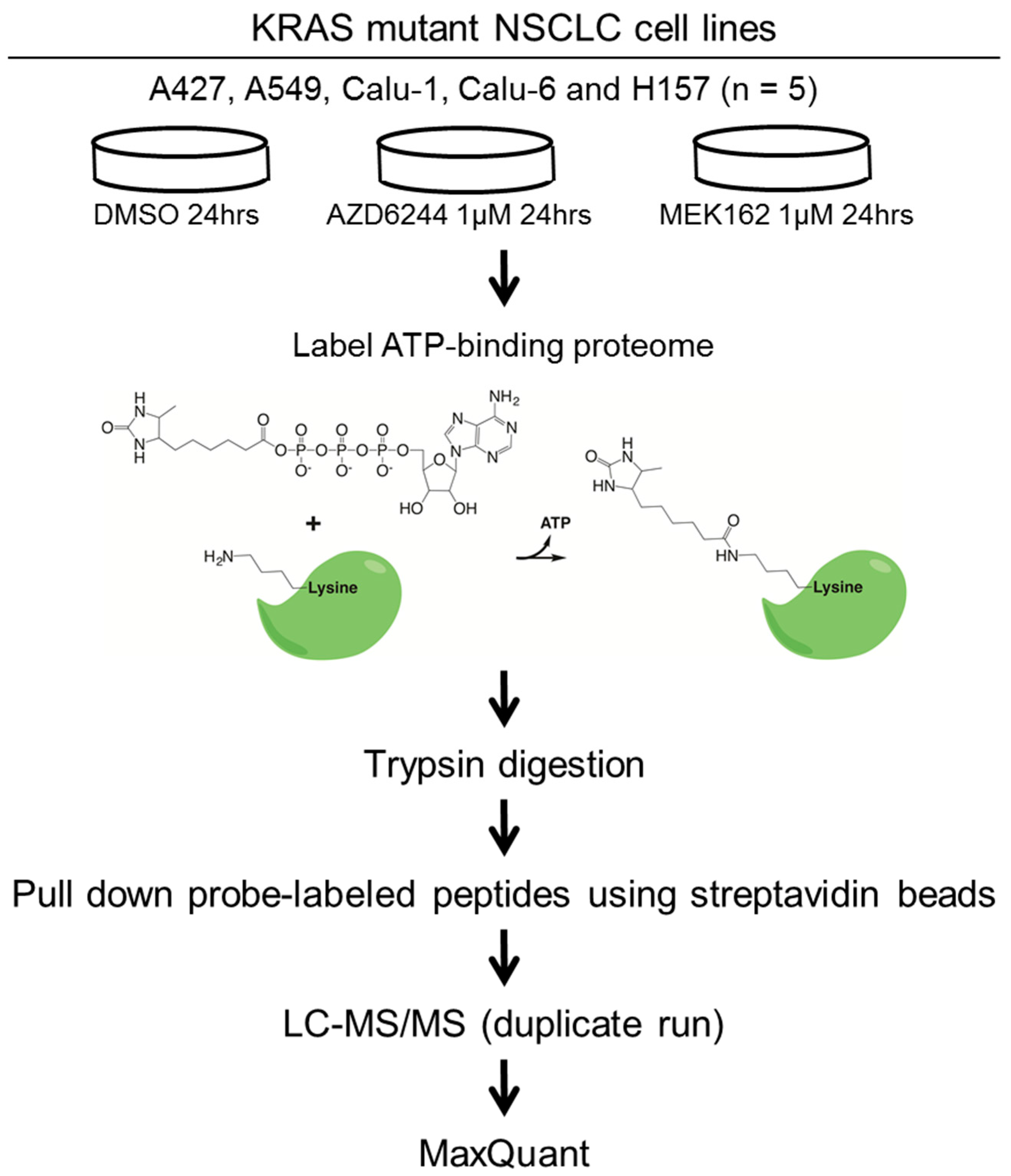
3. Materials and Methods
3.1. Cell Lines and Drugs
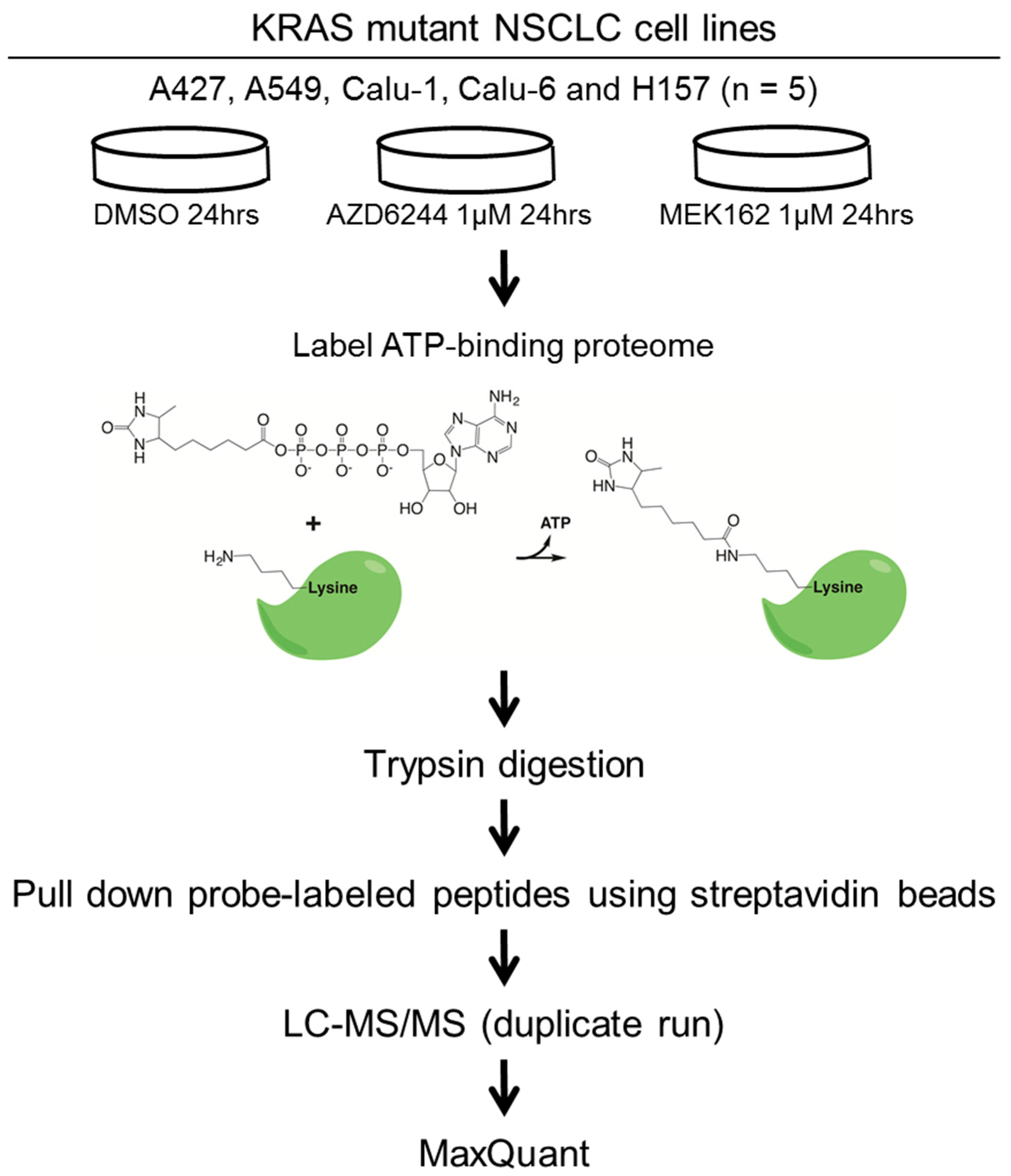
3.2. Drug Treatment and ATP Probe Labeling
3.3. MS Sample Preparation and LC-MS/MS Analysis
3.4. Data Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IRON | Iterative rank-order normalization |
| LC-MS/MS | Liquid chromatography/tandem mass spectrometry |
| NSCLC | Non-small cell lung cancer |
| PCA | Principal component analysis |
| SAPK | Stress-activated protein kinase |
References
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. Mtor inhibition induces upstream receptor tyrosine kinase signaling and activates akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of akt and eif4e survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. Akt inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. Mek inhibition leads to pi3k/akt activation by relieving a negative feedback on erbb receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of mek inhibition determines efficacy in mutant kras-versus braf-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of craf-mediated mek activation is required for effective mek inhibition in kras mutant tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012, 2, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Klinger, B.; Bluthgen, N. Consequences of feedback in signal transduction for targeted therapies. Biochem. Soc. Trans. 2014, 42, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted mek inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of lapatinib-induced kinome reprogramming in erbb2-positive breast cancer by targeting bet family bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.J.; Cox, N.J.; Zimmerman, E.I.; Dewar, B.J.; Duncan, J.S.; Whittle, M.C.; Nguyen, T.A.; Jones, L.S.; Ghose Roy, S.; Smalley, D.M.; et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE 2013, 8, e66755. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Kim, J.Y.; Watters, J.M.; Fang, B.; Kinose, F.; Song, L.; Koomen, J.M.; Teer, J.K.; Fisher, K.; Chen, Y.A.; et al. Adaptive responses to dasatinib-treated lung squamous cell cancer cells harboring ddr2 mutations. Cancer Res. 2014, 74, 7217–7228. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Zhang, G.; Smith, M.A.; Lopez, A.S.; Bai, Y.; Li, J.; Fang, B.; Koomen, J.; Rawal, B.; Fisher, K.J.; et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for t790m-related egfr-tki resistance in non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Welsh, E.A.; Oguz, U.; Fang, B.; Bai, Y.; Kinose, F.; Bronk, C.; Remsing Rix, L.L.; Beg, A.A.; Rix, U.; et al. Dissection of tbk1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Nomanbhoy, T.K.; Wu, J.; Brown, H.; Zhou, D.; Zhang, J.; Jagannathan, S.; Aban, A.; Okerberg, E.; Herring, C.; et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011, 18, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Trejo, C.L.; Juan, J.; Vicent, S.; Sweet-Cordero, A.; McMahon, M. Mek1/2 inhibition elicits regression of autochthonous lung tumors induced by krasg12d or brafv600e. Cancer Res. 2012, 72, 3048–3059. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Cohen, R.B.; Franklin, W.; Morris, C.; Wilson, D.; Molina, J.R.; Hanson, L.J.; Gore, L.; Chow, L.; Leong, S.; et al. Phase i pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor azd6244 (arry-142886) in patients with advanced cancers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for kras-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar] [CrossRef]
- Chen, Z.; Cheng, K.; Walton, Z.; Wang, Y.; Ebi, H.; Shimamura, T.; Liu, Y.; Tupper, T.; Ouyang, J.; Li, J.; et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012, 483, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of stk11 and tp53 with kras mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fang, B.; Kinose, F.; Bai, Y.; Kim, J.Y.; Chen, Y.A.; Rix, U.; Koomen, J.M.; Haura, E.B. Target identification in small cell lung cancer via integrated phenotypic screening and activity-based protein profiling. Mol. Cancer Ther. 2016, 15, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Guo, L.; Wang, Y. A targeted quantitative proteomics strategy for global kinome profiling of cancer cells and tissues. Mol. Cell. Proteom. 2014, 13, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Szardenings, A.K.; Liyanage, M.; Nomanbhoy, T.K.; Wu, M.; Weissig, H.; Aban, A.; Chun, D.; Tanner, S.; Kozarich, J.W. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry-Us 2007, 46, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of braf-mutant melanoma to braf inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Falck Miniotis, M.; Arunan, V.; Eykyn, T.R.; Marais, R.; Workman, P.; Leach, M.O.; Beloueche-Babari, M. Mek1/2 inhibition decreases lactate in braf-driven human cancer cells. Cancer Res. 2013, 73, 4039–4049. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Vultur, A.; Villanueva, J.; Krepler, C.; Rajan, G.; Chen, Q.; Xiao, M.; Li, L.; Gimotty, P.A.; Wilson, M.; Hayden, J.; et al. Mek inhibition affects stat3 signaling and invasion in human melanoma cell lines. Oncogene 2014, 33, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic rna interference reveals that oncogenic kras-driven cancers require tbk1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide rnai screen identifies multiple synthetic lethal interactions with the ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.M.; Chu, Q.S.; Leighl, N.; Laurie, S.A.; Fritsch, H.; Gaschler-Markefski, B.; Gyorffy, S.; Munzert, G. A phase i open-label dose-escalation study of intravenous bi 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer. Clin. Lung Cancer 2013, 14, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Reck, M.; Waller, C.F.; Kortsik, C.; Frickhofen, N.; Schuler, M.; Fritsch, H.; Gaschler-Markefski, B.; Hanft, G.; Munzert, G.; et al. The efficacy and safety of bi 2536, a novel plk-1 inhibitor, in patients with stage iiib/iv non-small cell lung cancer who had relapsed after, or failed, chemotherapy: Results from an open-label, randomized phase ii clinical trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2010, 5, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting er stress-induced autophagy overcomes braf inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Dudek-Peric, A.M.; Maes, H.; Garg, A.D.; Gabrysiak, M.; Demirsoy, S.; Swinnen, J.V.; Agostinis, P. Concurrent mek and autophagy inhibition is required to restore cell death associated danger-signalling in vemurafenib-resistant melanoma cells. Biochem. Pharm. 2015, 93, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Yue, P.; Zhang, G.; Owonikoko, T.K.; Khuri, F.R.; Sun, S.Y. Enhancing therapeutic efficacy of the mek inhibitor, mek162, by blocking autophagy or inhibiting pi3k/akt signaling in human lung cancer cells. Cancer Lett. 2015, 364, 70–78. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.E.; Niepel, M.; Haas, W.; Huttlin, E.; Sorger, P.K.; Gygi, S.P. Mass spectrometry based method to increase throughput for kinome analyses using atp probes. Anal. Chem. 2013, 85, 4666–4674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized rnai screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Bafna, S.; Giltnane, J.M.; Balko, J.M.; Estrada, M.V.; Meszoely, I.; Mayer, I.; Abramson, V.; Ye, F.; et al. Kinome-wide functional screen identifies role of plk1 in hormone-independent, er-positive breast cancer. Cancer Res. 2015, 75, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.M.; Garimella, S.V.; Jones, T.L.; Caplen, N.J.; Lipkowitz, S. Identification of wee1 as a potential molecular target in cancer cells by rnai screening of the human tyrosine kinome. Breast Cancer Res. Treat. 2010, 122, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Z.; Malysa, A.; Li, X.; Oliveira, P.; Zhang, Y.; Bepler, G. A kinome screen identifies checkpoint kinase 1 (chk1) as a sensitizer for rrm1-dependent gemcitabine efficacy. PLoS ONE 2013, 8, e58091. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Chartier, M.; Chenard, T.; Barker, J.; Najmanovich, R. Kinome render: A stand-alone and web-accessible tool to annotate the human protein kinome tree. PeerJ 2013, 1, e126. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Eschrich, S.A.; Hoerter, A.M. Libaffy: Software for processing affymetrix genechip data. Bioinformatics 2007, 23, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- Welsh, E.A.; Eschrich, S.A.; Berglund, A.E.; Fenstermacher, D.A. Iterative rank-order normalization of gene expression microarray data. BMC Bioinform. 2013, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.A.; Parapatics, K.; Welsh, E.A.; Müller, A.C.; Cao, H.; Fang, B.; Koomen, J.M.; Eschrich, S.A.; Bennett, K.L.; Haura, E.B. A pilot proteogenomic study with data integration identifies mct1 and glut1 as prognostic markers in lung adenocarcinoma. PLoS ONE 2015, 10, e0142162. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- RStudio Team. Rstudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Thomson Reuters sGenego Metacore. Available online: http//www.genego.com (accsessed on 29 December 2015).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A427 | p Value |
| LRRK2 in neurons in Parkinson‘s disease | 4.62 × 10−11 |
| Development_Slit-Robo signaling | 3.63 × 10−10 |
| Development_Regulation of cytoskeleton proteins | 2.76 × 10−9 |
| Cytoskeleton remodeling_Regulation of actin cytoskeleton by Rho GTPases | 1.53 × 10−8 |
| Regulation of CFTR activity (normal and CF) | 7.02 × 10−8 |
| A549 | p Value |
| LRRK2 in neurons in Parkinson‘s disease | 1.54 × 10−13 |
| Cytoskeleton remodeling_Cytoskeleton remodeling | 2.38 × 10−10 |
| Glycolysis and gluconeogenesis (short map) | 1.29 × 10−9 |
| Cytoskeleton remodeling_Hyaluronic acid/ CD44 signaling pathways | 4.43 × 10−8 |
| Cytoskeleton remodeling_TGF, WNT and cytoskeletal remodeling | 6.59 × 10−8 |
| Calu-1 | p Value |
| Glycolysis and gluconeogenesis (short map) | 7.76 × 10−7 |
| Regulation of degradation of deltaF508-CFTR in CF | 2.04 × 10−6 |
| Transcription_Role of Akt in hypoxia induced HIF1 activation | 3.03 × 10−6 |
| LRRK2 and immune function in Parkinson's disease | 1.88 × 10−5 |
| Glycolysis and gluconeogenesis p.3 | 2.96 × 10−5 |
| Calu-6 | p Value |
| Cytoskeleton remodeling_Cytoskeleton remodeling | 4.86 × 10−8 |
| CFTR folding and maturation (normal and CF) | 5.09 × 10−8 |
| Cell adhesion_PLAU signaling | 1.11 × 10−7 |
| Development_VEGF signaling via VEGFR2—generic cascades | 5.96 × 10−7 |
| Development_EGFR signaling pathway | 1.3 × 10−6 |
| H157 | p Value |
| LRRK2 in neurons in Parkinson‘s disease | 2.21 × 10−13 |
| Neurophysiological process_Receptor-mediated axon growth repulsion | 6.45 × 10−13 |
| Cytoskeleton remodeling_Cytoskeleton remodeling | 3.35 × 10−11 |
| Development_Slit-Robo signaling | 6.54 × 10−11 |
| Cytoskeleton remodeling_Regulation of actin cytoskeleton by Rho GTPases | 1.14 × 10−10 |
| Gene Symbol | Position | Cell Line | Direction of Change |
|---|---|---|---|
| ABL2 | 446 | A427, Calu-1 | Increase |
| AURKA | 258 | Calu-6, Calu-1, H157 | Decrease |
| CHUK | 146 | Calu-6, H157 | Decrease |
| CMPK1 | 16 | Calu-6, H157 | Decrease |
| CSNK2A2 | 159 | A427, H157 | Increase |
| JAK1 | 718 | A549, Calu-1 | Increase |
| LATS2 | 793 | A549, A427, Calu-1 | Increase |
| LATS2 | 697 | A549, A427 | Increase |
| MAP2K2 | 108 | A549, Calu-6 | Increase |
| MAP2K6 | 181 | Calu-6, A427 | Increase |
| MAST3 | 492 | A427, H157 | Increase |
| NADK2 | 76 | Calu-6, A427, Calu-1 | Increase |
| NEK2 | 143 | Calu-6, H157 | Decrease |
| NEK3 | 131 | Calu-6, H157 | Decrease |
| PGK1 | 184 | A549, A427 | Increase |
| PGK1 | 91 | A427, Calu-1 | Increase |
| PKM | 322 | A427, H157 | Increase |
| PKM | 66 | A549, A427 | Increase |
| PLK1 | 178 | A549, Calu-6, Calu-1 | Decrease |
| PLK1 | 82 | A549, H157 | Decrease |
| PRKAA1 | 40 | A427, Calu-1 | Increase |
| PRPF4B | 727 | A549, Calu-1 | Increase |
| ROCK2 | 1065 | A427, Calu-1 | Increase |
| STK38 | 118 | A427, Calu-1, H157 | Increase |
| STK38L | 119 | A427, Calu-1 | Increase |
| TK1 | 32 | Calu-6, Calu-1 | Decrease |
| TLK1 | 485 | Calu-6, Calu-1 | Increase |
| ULK1 | 140 | A549, Calu-1 | Increase |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-Y.; Stewart, P.A.; Borne, A.L.; Fang, B.; Welsh, E.A.; Chen, Y.A.; Eschrich, S.A.; Koomen, J.M.; Haura, E.B. Activity-Based Proteomics Reveals Heterogeneous Kinome and ATP-Binding Proteome Responses to MEK Inhibition in KRAS Mutant Lung Cancer. Proteomes 2016, 4, 16. https://doi.org/10.3390/proteomes4020016
Kim J-Y, Stewart PA, Borne AL, Fang B, Welsh EA, Chen YA, Eschrich SA, Koomen JM, Haura EB. Activity-Based Proteomics Reveals Heterogeneous Kinome and ATP-Binding Proteome Responses to MEK Inhibition in KRAS Mutant Lung Cancer. Proteomes. 2016; 4(2):16. https://doi.org/10.3390/proteomes4020016
Chicago/Turabian StyleKim, Jae-Young, Paul A. Stewart, Adam L. Borne, Bin Fang, Eric A. Welsh, Yian Ann Chen, Steven A. Eschrich, John M. Koomen, and Eric B. Haura. 2016. "Activity-Based Proteomics Reveals Heterogeneous Kinome and ATP-Binding Proteome Responses to MEK Inhibition in KRAS Mutant Lung Cancer" Proteomes 4, no. 2: 16. https://doi.org/10.3390/proteomes4020016
APA StyleKim, J.-Y., Stewart, P. A., Borne, A. L., Fang, B., Welsh, E. A., Chen, Y. A., Eschrich, S. A., Koomen, J. M., & Haura, E. B. (2016). Activity-Based Proteomics Reveals Heterogeneous Kinome and ATP-Binding Proteome Responses to MEK Inhibition in KRAS Mutant Lung Cancer. Proteomes, 4(2), 16. https://doi.org/10.3390/proteomes4020016