
Challenges and Strategies for Proteome Analysis of the Interaction of Human Pathogenic Fungi with Host Immune Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Immunoproteomics
2.1. Gel-Based Immunoproteomics
2.2. Gel-Free Immunoproteomics
3. Interaction of Macrophages with Human Pathogenic Fungi
3.1. Recognition of the Fungal Pathogen
3.2. Phagocytosis, Phagosomal Maturation and Killing of the Pathogen
3.3. Studying the Phagolysosomal Proteome
3.4. Current Methods for the Purification of Phagolysosomes
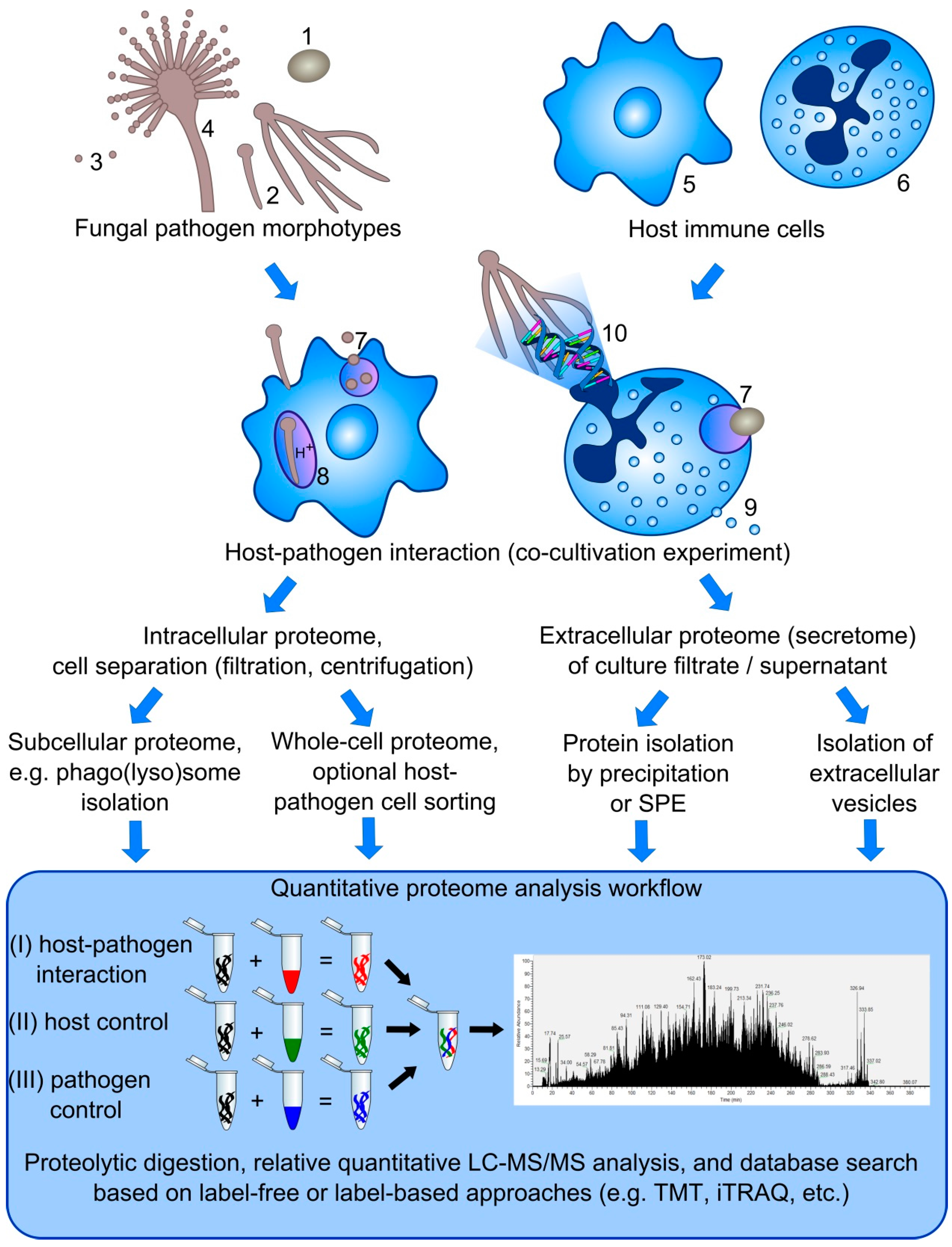
3.5. Extracellular Vesicles of Macrophages
4. Interaction of Human Pathogenic Fungi with Neutrophils
4.1. Neutrophil Phagocytosis and Phagosome Maturation
4.2. Neutrophil Extracellular Traps (NETs)
4.3. Proteome Analysis of Neutrophil-Pathogen Interactions
4.4. Studying the Phosphoproteome of Neutrophils: Protein Extraction Requirements
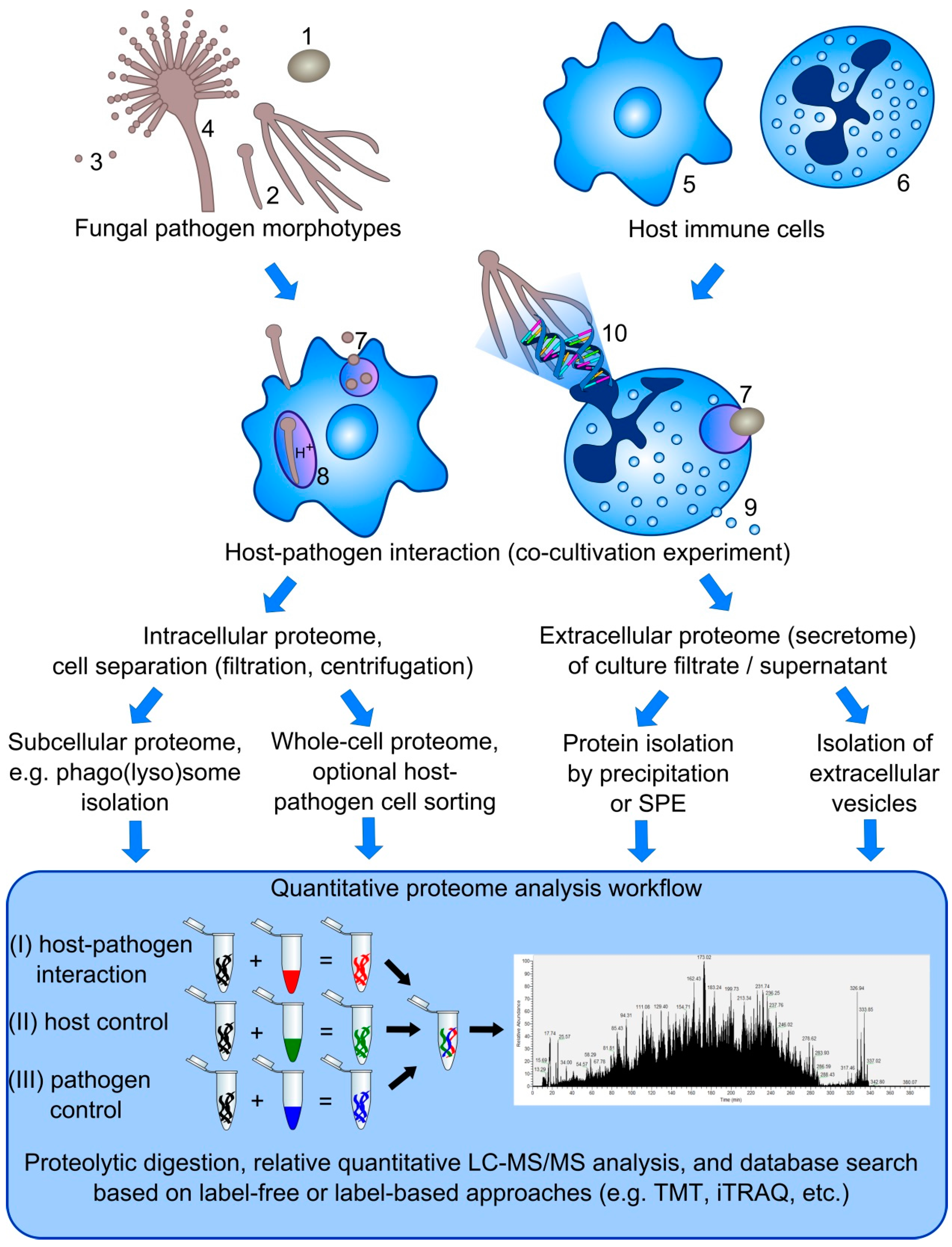
5. Challenges in the Simultaneous Dual Proteome Analysis of Host-Pathogen Interactions
5.1. Protein Complexity
5.2. Separation of Host Immune Cells from Fungal Pathogen Cells
5.3. Dynamic Range of Dual Proteome Studies
5.4. Quantitative Proteomics
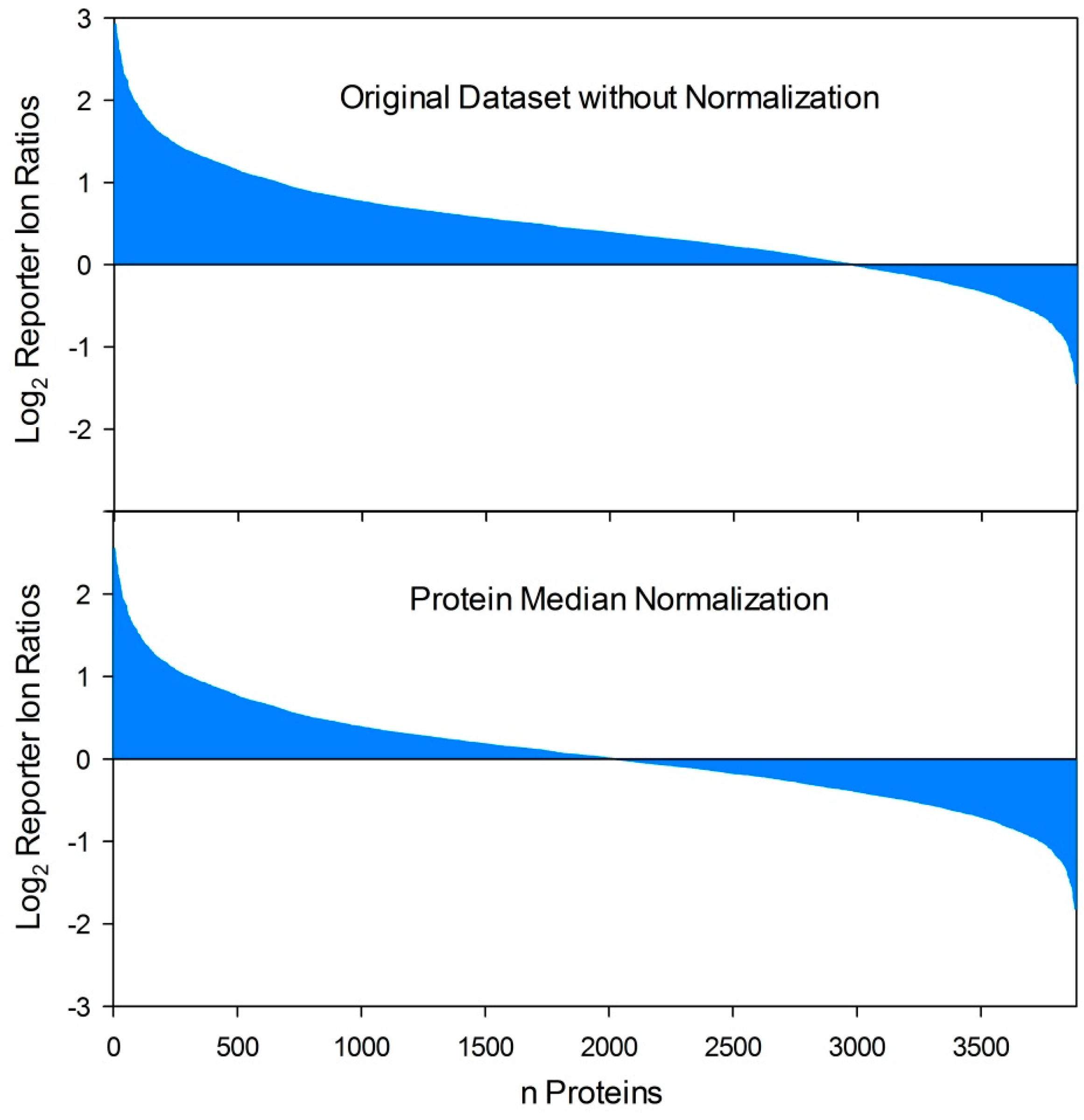
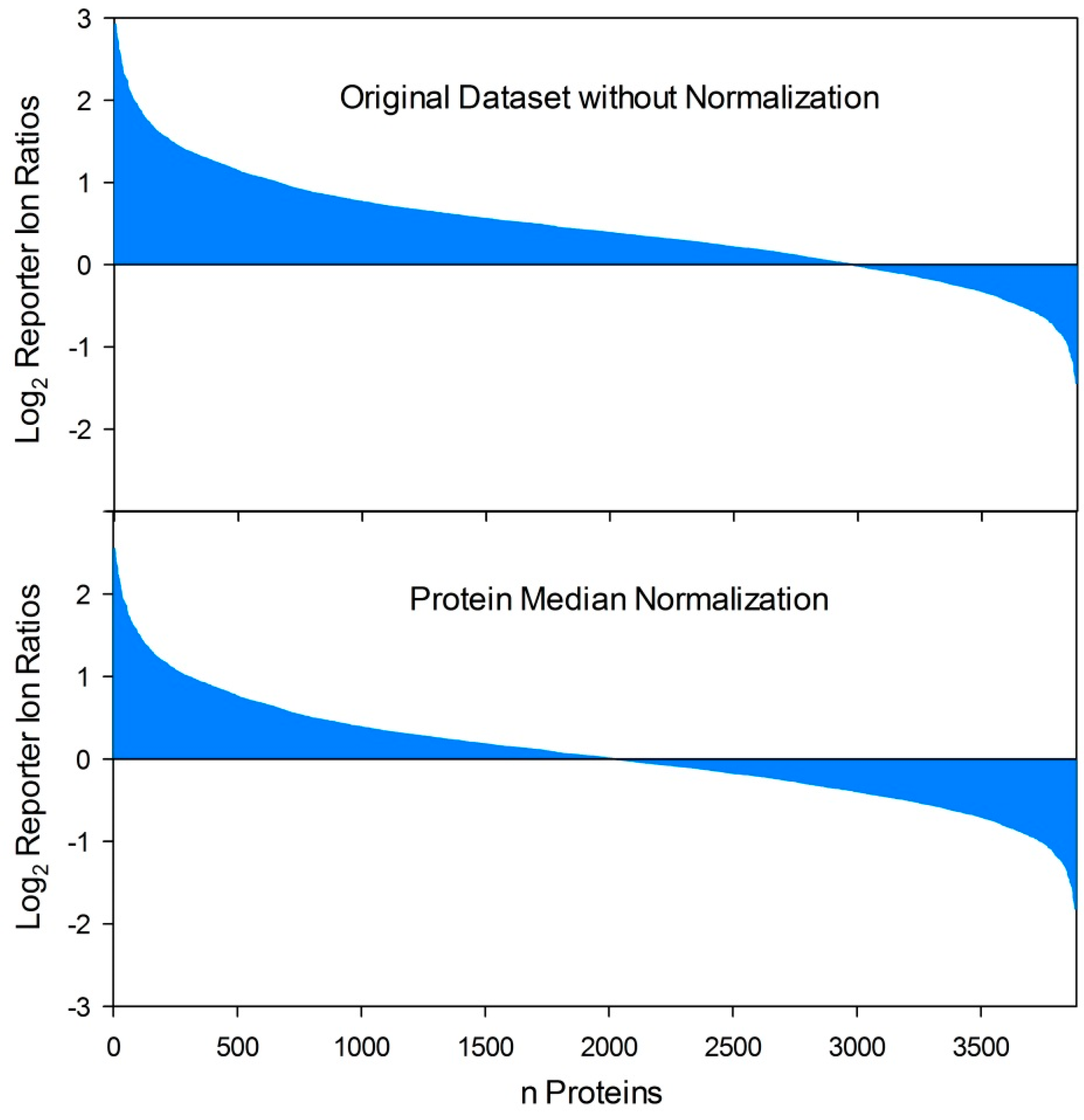
5.5. Normalization of Quantitative Dual Proteome Analysis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.R.; Casadevall, A.; Perfect, J. The spectrum of fungi that infects humans. Cold Spring Harb. Perspect. Med. 2015, 5, a019273. [Google Scholar] [CrossRef] [PubMed]
- Delaloye, J.; Calandra, T. Invasive candidiasis as a cause of sepsis in the critically ill patient. Virulence 2014, 5, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Papon, N.; Courdavault, V.; Clastre, M.; Bennett, R.J. Emerging and emerged pathogenic Candida species: Beyond the Candida albicans paradigm. PLoS Pathog. 2013, 9, e1003550. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Tekaia, F.; Latge, J.P. Aspergillus fumigatus: Saprophyte or pathogen? Curr. Opin. Microbiol. 2005, 8, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Warris, A. The biology of pulmonary aspergillus infections. J. Infect. 2014, 69 (Suppl. S1), S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, C.; Denning, D.W. The clinical spectrum of pulmonary aspergillosis. Thorax 2015, 70, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Balajee, S.A.; Kano, R.; Baddley, J.W.; Moser, S.A.; Marr, K.A.; Alexander, B.D.; Andes, D.; Kontoyiannis, D.P.; Perrone, G.; Peterson, S.; et al. Molecular identification of Aspergillus species collected for the transplant-associated infection surveillancenetwork. J. Clin. Microbiol. 2009, 47, 3138–3141. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Systemic fungal infections caused by Aspergillus species: Epidemiology, infection process and virulence determinants. Curr. Drug Targets 2005, 6, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Heinekamp, T.; Schmidt, H.; Lapp, K.; Pahtz, V.; Shopova, I.; Koster-Eiserfunke, N.; Kruger, T.; Kniemeyer, O.; Brakhage, A.A. Interference of Aspergillus fumigatus with the immune response. Semin. Immunopathol. 2015, 37, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Gullo, F.P.; Rossi, S.A.; Sardi Jde, C.; Teodoro, V.L.; Mendes-Giannini, M.J.; Fusco-Almeida, A.M. Cryptococcosis: Epidemiology, fungal resistance, and new alternatives for treatment. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Meyer, W.; Sorrell, T.C. Cryptococcus gattii infections. Clin. Microbiol. Rev. 2014, 27, 980–1024. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, T.R.; Alspaugh, J.A. The Cryptococcus neoformans capsule: A sword and a shield. Clin. Microbiol. Rev. 2012, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Feder, V.; Kmetzsch, L.; Staats, C.C.; Vidal-Figueiredo, N.; Ligabue-Braun, R.; Carlini, C.R.; Vainstein, M.H. Cryptococcus gattii urease as a virulence factor and the relevance of enzymatic activity in cryptococcosis pathogenesis. FEBS J. 2015, 282, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Mody, C.H. Cryptococcus. Proc. Am. Thorac. Soc. 2010, 7, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, F.; Wright, T.W. Pneumocystis: Where does it live? PLoS Pathog. 2012, 8, e1003025. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.F., Jr.; Limper, A.H. Current insights into the biology and pathogenesis of Pneumocystis pneumonia. Nat. Rev. Microbiol. 2007, 5, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Petrikkos, G.; Skiada, A.; Lortholary, O.; Roilides, E.; Walsh, T.J.; Kontoyiannis, D.P. Epidemiology and clinical manifestations of mucormycosis. Clin. Infect. Dis. 2012, 54 (Suppl. S1), S23–S34. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.S.; Tebbets, B. Dimorphism and virulence in fungi. Curr. Opin. Microbiol. 2007, 10, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Rappleye, C.A. Histoplasma mechanisms of pathogenesis—One portfolio doesn’t fit all. FEMS Microbiol. Lett. 2011, 324, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A.; Bruns, S.; Thywissen, A.; Zipfel, P.F.; Behnsen, J. Interaction of phagocytes with filamentous fungi. Curr. Opin. Microbiol. 2010, 13, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.K.; Levitz, S.M. Interactions of fungi with phagocytes. Curr. Opin. Microbiol. 2002, 5, 359–365. [Google Scholar] [CrossRef]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2011, 11, 275–288. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.; Heesemann, L.; Wagener, J.; Marcos, V.; Hartl, D.; Loeffler, J.; Heesemann, J.; Ebel, F. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. 2010, 12, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywissen, A.; Jeron, A.; Latge, J.P.; Brakhage, A.A.; Gunzer, M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010, 6, e1000873. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ortiz, Z.G.; Means, T.K. The role of dendritic cells in the innate recognition of pathogenic fungi (A. fumigatus, C. neoformans and C. albicans). Virulence 2012, 3, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Lass-Florl, C.; Roilides, E.; Loffler, J.; Wilflingseder, D.; Romani, L. Minireview: Host defence in invasive aspergillosis. Mycoses 2013, 56, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.; Minuzzi, F.; Bignell, E. The host-infecting fungal transcriptome. FEMS Microbiol. Lett. 2010, 307, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.C.; Bender, J.A.; Fink, G.R. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 2004, 3, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Thewes, S.; Zakikhany, K.; Fradin, C.; Albrecht, A.; Almeida, R.; Brunke, S.; Grosse, K.; Martin, R.; Mayer, F.; et al. Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res. 2009, 9, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Sugui, J.A.; Kim, H.S.; Zarember, K.A.; Chang, Y.C.; Gallin, J.I.; Nierman, W.C.; Kwon-Chung, K.J. Genes differentially expressed in conidia and hyphae of Aspergillus fumigatus upon exposure to human neutrophils. PLoS ONE 2008, 3, e2655. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.O.; Varga, J.J.; Hornbach, A.; Mezger, M.; Sennefelder, H.; Kneitz, S.; Kurzai, O.; Krappmann, S.; Einsele, H.; Nierman, W.C.; et al. The temporal dynamics of differential gene expression in Aspergillus fumigatus interacting with human immature dendritic cells in vitro. PLoS ONE 2011, 6, e16016. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, T.; Toth, A.; Hamari, Z.; Falus, A.; Eder, K.; Vagvolgyi, C.; Guimaraes, A.J.; Nosanchuk, J.D.; Gacser, A. Transcriptome profile of the murine macrophage cell response to Candida parapsilosis. Fungal Genet. Biol. 2014, 65, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fradin, C.; Mavor, A.L.; Weindl, G.; Schaller, M.; Hanke, K.; Kaufmann, S.H.; Mollenkopf, H.; Hube, B. The early transcriptional response of human granulocytes to infection with Candida albicans is not essential for killing but reflects cellular communications. Infect. Immun. 2007, 75, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, C.; Jia, X.; Wang, S.; Wang, J.; Chen, Y.; Zhao, J.; Tian, S.; Han, X.; Han, L. Transcriptome Profiles of Human Lung Epithelial Cells A549 Interacting with Aspergillus fumigatus by RNA-Seq. PLoS ONE 2015, 10, e0135720. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, J.L.; Gomez, P.; Ruan, J.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS ONE 2011, 6, e20527. [Google Scholar] [CrossRef] [PubMed]
- Gomez, P.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Functional genomics of human bronchial epithelial cells directly interacting with conidia of Aspergillus fumigatus. BMC Genom. 2010, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Zakikhany, K.; Naglik, J.R.; Schmidt-Westhausen, A.; Holland, G.; Schaller, M.; Hube, B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol. 2007, 9, 2938–2954. [Google Scholar] [CrossRef] [PubMed]
- Barker, K.S.; Park, H.; Phan, Q.T.; Xu, L.; Homayouni, R.; Rogers, P.D.; Filler, S.G. Transcriptome profile of the vascular endothelial cell response to Candida albicans. J. Infect. Dis. 2008, 198, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Fekkar, A.; Balloy, V.; Pionneau, C.; Marinach-Patrice, C.; Chignard, M.; Mazier, D. Secretome of human bronchial epithelial cells in response to the fungal pathogen Aspergillus fumigatus analyzed by differential in-gel electrophoresis. J. Infect. Dis. 2012, 205, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Curty, N.; Kubitschek-Barreira, P.H.; Neves, G.W.; Gomes, D.; Pizzatti, L.; Abdelhay, E.; Souza, G.H.M.F.; Lopes-Bezerra, L.M. Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J. Proteom. 2014, 97, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, P.R.; Grabher, G.; Stoffler, G. Comprehensive detection of immunorelevant Borrelia garinii antigens by two-dimensional electrophoresis. Electrophoresis 1999, 20, 3611–3622. [Google Scholar] [CrossRef]
- Jungblut, P.R. Proteome analysis of bacterial pathogens. Microbes Infect. 2001, 3, 831–840. [Google Scholar] [CrossRef]
- Klade, C.S.; Voss, T.; Krystek, E.; Ahorn, H.; Zatloukal, K.; Pummer, K.; Adolf, G.R. Identification of tumor antigens in renal cell carcinoma by serological proteome analysis. Proteomics 2001, 1, 890–898. [Google Scholar] [CrossRef]
- Pitarch, A.; Abian, J.; Carrascal, M.; Sanchez, M.; Nombela, C.; Gil, C. Proteomics-based identification of novel Candida albicans antigens for diagnosis of systemic candidiasis in patients with underlying hematological malignancies. Proteomics 2004, 4, 3084–3106. [Google Scholar] [CrossRef] [PubMed]
- Pitarch, A.; Jimenez, A.; Nombela, C.; Gil, C. Decoding serological response to Candida cell wall immunome into novel diagnostic, prognostic, and therapeutic candidates for systemic candidiasis by proteomic and bioinformatic analyses. Mol. Cell Proteom. 2006, 5, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Pitarch, A.; Nombela, C.; Gil, C. Serum antibody signature directed against Candida albicans Hsp90 and enolase detects invasive candidiasis in non-neutropenic patients. J. Proteome Res. 2014, 13, 5165–5184. [Google Scholar] [CrossRef] [PubMed]
- Pitarch, A.; Nombela, C.; Gil, C. Seroprofiling at the Candida albicans protein species level unveils an accurate molecular discriminator for candidemia. J. Proteomics 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.G.; Kirsch, A.I.; Zeller, S.; Menz, G.; Rhyner, C.; Crameri, R. Molecular and immunological characterization of Asp f 34, a novel major cell wall allergen of Aspergillus fumigatus. Allergy 2009, 64, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.N.; Li, F.Q.; Lu, J.F.; Kong, X.X.; Wang, S.Q.; Huang, M.; Shao, H.F.; Shao, S.H. Antibody specific to thioredoxin reductase as a new biomarker for serodiagnosis of invasive aspergillosis in non-neutropenic patients. Clin. Chim. Acta 2012, 413, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H. Antigen arrays for antibody profiling. Curr. Opin. Chem. Biol. 2006, 10, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Robinson, W.H. Proteomic biomarkers for autoimmune disease. Proteomics 2006, 6, 4100–4105. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; DiGennaro, C.; Hueber, W.; Haab, B.B.; Kamachi, M.; Dean, E.J.; Fournel, S.; Fong, D.; Genovese, M.C.; de Vegvar, H.E.; et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 2002, 8, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Mochon, A.B.; Jin, Y.; Kayala, M.A.; Wingard, J.R.; Clancy, C.J.; Nguyen, M.H.; Felgner, P.; Baldi, P.; Liu, H. Serological profiling of a Candida albicans protein microarray reveals permanent host-pathogen interplay and stage-specific responses during candidemia. PLoS Pathog. 2010, 6, e1000827. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Schaeps, R.M.; Swinkels, D.W. Immunoproteomics: From biomarker discovery to diagnostic applications. Proteom. Clin. Appl. 2008, 2, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A. Macrophages in the embryo and beyond: Much more than just giant phagocytes. Genesis 2008, 46, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Seider, K.; Heyken, A.; Luttich, A.; Miramon, P.; Hube, B. Interaction of pathogenic yeasts with phagocytes: Survival, persistence and escape. Curr. Opin. Microbiol. 2010, 13, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, K.; Jahn, B.; Gehringer, H.; Schmidt, A.; Wanner, G.; Brakhage, A.A. Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med. Microbiol. Immunol. 1998, 187, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Thywissen, A.; Heinekamp, T.; Dahse, H.M.; Schmaler-Ripcke, J.; Nietzsche, S.; Zipfel, P.F.; Brakhage, A.A. Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis Pathway. Front. Microbiol. 2011, 2, 96. [Google Scholar] [CrossRef] [PubMed]
- Luther, K.; Torosantucci, A.; Brakhage, A.A.; Heesemann, J.; Ebel, F. Phagocytosis of Aspergillus fumigatus conidia by murine macrophages involves recognition by the dectin-1 β-glucan receptor and Toll-like receptor 2. Cell Microbiol. 2007, 9, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, T.R.; Giles, S.S.; Aimanianda, V.; Latge, J.P.; Hull, C.M.; Keller, N.P. Aspergillus fumigatus LaeA-mediated phagocytosis is associated with a decreased hydrophobin layer. Infect. Immun. 2010, 78, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Levitz, S.M. Innate recognition of fungal cell walls. PLoS Pathog. 2010, 6, e1000758. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.T.; Fink, G.R. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodas, R.; Zaragoza, O. Catch me if you can: Phagocytosis and killing avoidance by Cryptococcus neoformans. FEMS Immunol. Med. Microbiol. 2012, 64, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.; Latge, J.P. Aspergillus fumigatus cell wall: Composition and biosynthesis. Med. Mycol. 2001, 39 (Suppl. S1), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, J.; Elorza, M.V.; Valentin, E.; Sentandreu, R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006, 6, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Gersuk, G.M.; Underhill, D.M.; Zhu, L.; Marr, K.A. Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J. Immunol. 2006, 176, 3717–3724. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Metz, A.E.; Horn, D.; Schoeb, T.R.; Hewitt, M.M.; Schwiebert, L.M.; Faro-Trindade, I.; Brown, G.D.; Steele, C. Requisite role for the dectin-1 β-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 2009, 182, 4938–4946. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Kirschning, C.J.; Nikolaus, T.; Wagner, H.; Heesemann, J.; Ebel, F. Toll-like receptor (TLR) 2 and TLR4 are essential for Aspergillus-induced activation of murine macrophages. Cell Microbiol. 2003, 5, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Warris, A.; van der Meer, J.W.; Fenton, M.J.; Verver-Janssen, T.J.; Jacobs, L.E.; Andresen, T.; Verweij, P.E.; Kullberg, B.J. Aspergillus fumigatus evades immune recognition during germination through loss of toll-like receptor-4-mediated signal transduction. J. Infect. Dis. 2003, 188, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, D.; Dominguez-Soto, A.; Ancochea, J.; Jimenez-Heffernan, J.A.; Leal, J.A.; Corbi, A.L. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin mediates binding and internalization of Aspergillus fumigatus conidia by dendritic cells and macrophages. J. Immunol. 2004, 173, 5635–5643. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, S.; Montagnoli, C.; Bozza, S.; Gaziano, R.; Rossi, G.; Mambula, S.S.; Vecchi, A.; Mantovani, A.; Levitz, S.M.; Romani, L. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J. Immunol. 2004, 172, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.; Rapaka, R.R.; Metz, A.; Pop, S.M.; Williams, D.L.; Gordon, S.; Kolls, J.K.; Brown, G.D. The β-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.Y.; Vonk, A.G.; Kullberg, B.J.; Verweij, P.E.; Verschueren, I.; van der Meer, J.W.; Joosten, L.A.; Latge, J.P.; Netea, M.G. Aspergillus fumigatus cell wall components differentially modulate host TLR2 and TLR4 responses. Microbes Infect. 2011, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sukhithasri, V.; Nisha, N.; Biswas, L.; Anil Kumar, V.; Biswas, R. Innate immune recognition of microbial cell wall components and microbial strategies to evade such recognitions. Microbiol. Res. 2013, 168, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Ozinsky, A. Phagocytosis of microbes: Complexity in action. Annu. Rev. Immunol. 2002, 20, 825–852. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Granet, O.; Philippe, B.; Boleti, H.; Boisvieux-Ulrich, E.; Grenet, D.; Stern, M.; Latge, J.P. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 2003, 71, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M. ER-mediated phagocytosis: A new membrane for new functions. Nat. Rev. Immunol. 2003, 3, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Philippe, B.; Ibrahim-Granet, O.; Prevost, M.C.; Gougerot-Pocidalo, M.A.; Sanchez Perez, M.; van der Meeren, A.; Latge, J.P. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect. Immun. 2003, 71, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Arenas, E.; Bleck, C.K.; Nombela, C.; Gil, C.; Griffiths, G.; Diez-Orejas, R. Candida albicans actively modulates intracellular membrane trafficking in mouse macrophage phagosomes. Cell Microbiol. 2009, 11, 560–589. [Google Scholar] [CrossRef] [PubMed]
- Haverland, N.A.; Fox, H.S.; Ciborowski, P. Quantitative proteomics by SWATH-MS reveals altered expression of nucleic acid binding and regulatory proteins in HIV-1-infected macrophages. J. Proteome Res. 2014, 13, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Stone, D.K.; Yu, F.; Zeng, Y.; Gendelman, H.E. Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage. J. Proteome Res. 2010, 9, 6759–6773. [Google Scholar] [CrossRef] [PubMed]
- Lietzen, N.; Ohman, T.; Rintahaka, J.; Julkunen, I.; Aittokallio, T.; Matikainen, S.; Nyman, T.A. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 2011, 7, e1001340. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Bai, J.; Zhang, L.; Liu, J.; Jiang, Z.; Michal, J.J.; He, Q.; Jiang, P. Two-dimensional liquid chromatography-tandem mass spectrometry coupled with isobaric tags for relative and absolute quantification (iTRAQ) labeling approach revealed first proteome profiles of pulmonary alveolar macrophages infected with porcine reproductive and respiratory syndrome virus. J. Proteome Res. 2012, 11, 2890–2903. [Google Scholar] [PubMed]
- Luo, R.; Fang, L.; Jin, H.; Wang, D.; An, K.; Xu, N.; Chen, H.; Xiao, S. Label-free quantitative phosphoproteomic analysis reveals differentially regulated proteins and pathway in PRRSV-infected pulmonary alveolar macrophages. J. Proteome Res. 2014, 13, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Depke, M.; Breitbach, K.; Dinh Hoang Dang, K.; Brinkmann, L.; Salazar, M.G.; Dhople, V.M.; Bast, A.; Steil, L.; Schmidt, F.; Steinmetz, I.; et al. Bone marrow-derived macrophages from BALB/c and C57BL/6 mice fundamentally differ in their respiratory chain complex proteins, lysosomal enzymes and components of antioxidant stress systems. J. Proteom. 2014, 103, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Dreisbach, A.; Otto, A.; Becher, D.; Bernhardt, J.; Hecker, M.; Peppelenbosch, M.P.; van Dijl, J.M. Mapping of interactions between human macrophages and Staphylococcus aureus reveals an involvement of MAP kinase signaling in the host defense. J. Proteome Res. 2011, 10, 4018–4032. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, D.; Tang, S.; Chen, X.; Gao, Q. PPE38 of Mycobacterium marinum triggers the cross-talk of multiple pathways involved in the host response, as revealed by subcellular quantitative proteomics. J. Proteome Res. 2013, 12, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Midha, M.K.; Verma, H.N.; Basu, A.; Rao, K.V.; Manivel, V. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci. Rep. 2013, 3, 1328. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, W.; Liao, G.; Xie, J. Mycobacterium tuberculosis-specific phagosome proteome and underlying signaling pathways. J. Proteome Res. 2012, 11, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Solano, L.; Nombela, C.; Molero, G.; Gil, C. Differential protein expression of murine macrophages upon interaction with Candida albicans. Proteomics 2006, 6 (Suppl. S1), S133–S144. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Solano, L.; Reales-Calderon, J.A.; Nombela, C.; Molero, G.; Gil, C. Proteomics of RAW 264.7 macrophages upon interaction with heat-inactivated Candida albicans cells unravel an anti-inflammatory response. Proteomics 2009, 9, 2995–3010. [Google Scholar] [CrossRef] [PubMed]
- Reales-Calderon, J.A.; Martinez-Solano, L.; Martinez-Gomariz, M.; Nombela, C.; Molero, G.; Gil, C. Sub-proteomic study on macrophage response to Candida albicans unravels new proteins involved in the host defense against the fungus. J. Proteom. 2012, 75, 4734–4746. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Arenas, E.; Cabezon, V.; Bermejo, C.; Arroyo, J.; Nombela, C.; Diez-Orejas, R.; Gil, C. Integrated proteomics and genomics strategies bring new insight into Candida albicans response upon macrophage interaction. Mol. Cell Proteom. 2007, 6, 460–478. [Google Scholar] [CrossRef] [PubMed]
- Vialas, V.; Nogales-Cadenas, R.; Nombela, C.; Pascual-Montano, A.; Gil, C. Proteopathogen, a protein database for studying Candida albicans—Host interaction. Proteomics 2009, 9, 4664–4668. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.; Vaz, C.; Carvalho-Pereira, J.; Pais, C.; Sampaio, P. A new method for yeast phagocytosis analysis by flow cytometry. J. Microbiol. Methods 2014, 101, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Kim, K.Y.; Paik, Y.K. Alterations of protein expression in macrophages in response to Candida albicans infection. Mol. Cells 2005, 20, 271–279. [Google Scholar] [PubMed]
- Reales-Calderon, J.A.; Sylvester, M.; Strijbis, K.; Jensen, O.N.; Nombela, C.; Molero, G.; Gil, C. Candida albicans induces pro-inflammatory and anti-apoptotic signals in macrophages as revealed by quantitative proteomics and phosphoproteomics. J. Proteom. 2013, 91, 106–135. [Google Scholar] [CrossRef] [PubMed]
- Reales-Calderon, J.A.; Aguilera-Montilla, N.; Corbi, A.L.; Molero, G.; Gil, C. Proteomic characterization of human proinflammatory M1 and anti-inflammatory M2 macrophages and their response to Candida albicans. Proteomics 2014, 14, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Huber, L.A.; Parton, R.G.; Griffiths, G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 1994, 124, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Jethwaney, D.; Schilling, B.; Clemens, D.L.; Gibson, B.W.; Horwitz, M.A. The Mycobacterium bovis bacille Calmette-Guerin phagosome proteome. Mol. Cell Proteom. 2010, 9, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Urwyler, S.; Brombacher, E.; Hilbi, H. Endosomal and secretory markers of the Legionella-containing vacuole. Commun. Integr. Biol. 2009, 2, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Finsel, I.; Otto, A.; Pfaffinger, G.; Rothmeier, E.; Hecker, M.; Becher, D.; Hilbi, H. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell Microbiol. 2014, 16, 1034–1052. [Google Scholar] [PubMed]
- Steinhauser, C.; Heigl, U.; Tchikov, V.; Schwarz, J.; Gutsmann, T.; Seeger, K.; Brandenburg, J.; Fritsch, J.; Schroeder, J.; Wiesmuller, K.H.; et al. Lipid-labeling facilitates a novel magnetic isolation procedure to characterize pathogen-containing phagosomes. Traffic 2013, 14, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Cypryk, W.; Ohman, T.; Eskelinen, E.L.; Matikainen, S.; Nyman, T.A. Quantitative proteomics of extracellular vesicles released from human monocyte-derived macrophages upon β-glucan stimulation. J. Proteome Res. 2014, 13, 2468–2477. [Google Scholar] [CrossRef] [PubMed]
- Bonnett, C.R.; Cornish, E.J.; Harmsen, A.G.; Burritt, J.B. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infect. Immun. 2006, 74, 6528–6539. [Google Scholar] [CrossRef] [PubMed]
- Feldmesser, M. Role of neutrophils in invasive aspergillosis. Infect Immun 2006, 74, 6514–6516. [Google Scholar] [CrossRef] [PubMed]
- Mircescu, M.M.; Lipuma, L.; van Rooijen, N.; Pamer, E.G.; Hohl, T.M. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J. Infect. Dis. 2009, 200, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Stephens-Romero, S.D.; Mednick, A.J.; Feldmesser, M. The pathogenesis of fatal outcome in murine pulmonary aspergillosis depends on the neutrophil depletion strategy. Infect. Immun. 2005, 73, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.D.; Williams, D.A.; Gifford, M.A.; Li, L.L.; Du, X.; Fisherman, J.; Orkin, S.H.; Doerschuk, C.M.; Dinauer, M.C. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet. 1995, 9, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.B.; Stephens, L.R.; Hawkins, P.T. Activation of the neutrophil NADPH oxidase by Aspergillus fumigatus. Ann. N. Y. Acad. Sci. 2012, 1273, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.F.; Chang, Y.C.; Washburn, R.G.; Wheeler, M.H.; Kwon-Chung, K.J. The developmentally regulated alb1 gene of Aspergillus fumigatus: Its role in modulation of conidial morphology and virulence. J. Bacteriol. 1998, 180, 3031–3038. [Google Scholar] [PubMed]
- Segal, A.W.; Coade, S.B. Kinetics of oxygen consumption by phagocytosing human neutrophils. Biochem. Biophys. Res. Commun. 1978, 84, 611–617. [Google Scholar] [CrossRef]
- Xu, P.; Crawford, M.; Way, M.; Godovac-Zimmermann, J.; Segal, A.W.; Radulovic, M. Subproteome analysis of the neutrophil cytoskeleton. Proteomics 2009, 9, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.D.; Nascimento, M.T.; Decote-Ricardo, D.; Corte-Real, S.; Morrot, A.; Heise, N.; Nunes, M.P.; Previato, J.O.; Mendonca-Previato, L.; DosReis, G.A.; et al. Capsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci. Rep. 2015, 5, 8008. [Google Scholar] [CrossRef] [PubMed]
- Mejia, S.P.; Cano, L.E.; Lopez, J.A.; Hernandez, O.; Gonzalez, A. Human neutrophils produce extracellular traps against Paracoccidioides brasiliensis. Microbiology 2015, 161, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Frealle, E.; Aliouat-Denis, C.M.; Delhaes, L.; Hot, D.; Dei-Cas, E. Transcriptomic insights into the oxidative response of stress-exposed Aspergillus fumigatus. Curr. Pharm. Des. 2013, 19, 3713–3737. [Google Scholar] [CrossRef] [PubMed]
- Hunniger, K.; Lehnert, T.; Bieber, K.; Martin, R.; Figge, M.T.; Kurzai, O. A virtual infection model quantifies innate effector mechanisms and Candida albicans immune escape in human blood. PLoS Comput. Biol. 2014, 10, e1003479. [Google Scholar] [CrossRef] [PubMed]
- Chevallet, M.; Diemer, H.; van Dorssealer, A.; Villiers, C.; Rabilloud, T. Toward a better analysis of secreted proteins: The example of the myeloid cells secretome. Proteomics 2007, 7, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Aryal, U.K.; Dai, Z.; Mason, A.C.; Monroe, M.E.; Tian, Z.X.; Zhou, J.Y.; Su, D.; Weitz, K.K.; Liu, T.; et al. Mapping N-linked glycosylation sites in the secretome and whole cells of Aspergillus niger using hydrazide chemistry and mass spectrometry. J. Proteome Res. 2012, 11, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, K.; Winter, M.; Berriel Diaz, M.; Herzig, S.; Krijgsveld, J. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 2012, 30, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.J.; Jin, Y.; Knudsen, G.M.; Perera, N.C.; Jenne, D.E.; Murphy, J.E.; Craik, C.S.; Hermiston, T.W. Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS ONE 2013, 8, e75141. [Google Scholar] [CrossRef] [PubMed]
- De Souza Castro, M.; de Sa, N.M.; Gadelha, R.P.; de Sousa, M.V.; Ricart, C.A.; Fontes, B.; Fontes, W. Proteome analysis of resting human neutrophils. Protein Pept. Lett. 2006, 13, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Tomazella, G.G.; da Silva, I.; Laure, H.J.; Rosa, J.C.; Chammas, R.; Wiker, H.G.; de Souza, G.A.; Greene, L.J. Proteomic analysis of total cellular proteins of human neutrophils. Proteome Sci. 2009, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Tomazella, G.G.; daSilva, I.; Thome, C.H.; Greene, L.J.; Koehler, C.J.; Thiede, B.; Wiker, H.G.; de Souza, G.A. Analysis of detergent-insoluble and whole cell lysate fractions of resting neutrophils using high-resolution mass spectrometry. J. Proteome Res. 2010, 9, 2030–2036. [Google Scholar] [CrossRef] [PubMed]
- Eitzen, G.; Lo, A.N.; Mitchell, T.; Kim, J.D.; Chao, D.V.; Lacy, P. Proteomic analysis of secretagogue-stimulated neutrophils implicates a role for actin and actin-interacting proteins in Rac2-mediated granule exocytosis. Proteome Sci. 2011, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Rorvig, S.; Ostergaard, O.; Heegaard, N.H.; Borregaard, N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: Correlation with transcriptome profiling of neutrophil precursors. J. Leukoc. Biol. 2013, 94, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Al-Shami, A.; Gilbert, C.; Barabe, F.; Gaudry, M.; Naccache, P.H. Preservation of the pattern of tyrosine phosphorylation in human neutrophil lysates. J. Immunol. Methods 1997, 202, 183–191. [Google Scholar] [CrossRef]
- Gilbert, C.; Rollet-Labelle, E.; Naccache, P.H. Preservation of the pattern of tyrosine phosphorylation in human neutrophil lysates. II. A sequential lysis protocol for the analysis of tyrosine phosphorylation-dependent signalling. J. Immunol. Methods 2002, 261, 85–101. [Google Scholar] [CrossRef]
- Yan, S.R.; Fumagalli, L.; Berton, G. Activation of SRC family kinases in human neutrophils. Evidence that p58C-FGR and p53/56LYN redistributed to a Triton X-100-insoluble cytoskeletal fraction, also enriched in the caveolar protein caveolin, display an enhanced kinase activity. FEBS Lett. 1996, 380, 198–203. [Google Scholar] [PubMed]
- Jones, T.; Federspiel, N.A.; Chibana, H.; Dungan, J.; Kalman, S.; Magee, B.B.; Newport, G.; Thorstenson, Y.R.; Agabian, N.; Magee, P.T.; et al. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. USA 2004, 101, 7329–7334. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.R.; van Het Hoog, M.; D’Enfert, C.; Martchenko, M.; Dungan, J.; Kuo, A.; Inglis, D.O.; Uhl, M.A.; Hogues, H.; Berriman, M.; et al. A human-curated annotation of the Candida albicans genome. PLoS Genet. 2005, 1, 36–57. [Google Scholar] [CrossRef] [PubMed]
- Nierman, W.C.; Pain, A.; Anderson, M.J.; Wortman, J.R.; Kim, H.S.; Arroyo, J.; Berriman, M.; Abe, K.; Archer, D.B.; Bermejo, C.; et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 2005, 438, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Pichler, P.; Swart, R.; Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanoLC-MS/MS using ultralong gradients. Nat. Protoc. 2012, 7, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Moruz, L.; Pichler, P.; Stranzl, T.; Mechtler, K.; Kall, L. Optimized nonlinear gradients for reversed-phase liquid chromatography in shotgun proteomics. Anal. Chem. 2013, 85, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Karimpour-Fard, A.; Epperson, L.E.; Hunter, L.E. A survey of computational tools for downstream analysis of proteomic and other omic datasets. Hum. Genom. 2015, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Zollner, A.; Maier, D.; Albermann, K.; Hani, J.; Mokrejs, M.; Tetko, I.; Guldener, U.; Mannhaupt, G.; Munsterkotter, M.; et al. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucl. Acids Res. 2004, 32, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucl. Acids Res 2013, 41, D793–D800. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Carlson, A.; Sinitcyn, P.; Mann, M.; Cox, J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Junker, J.; Bielow, C.; Bertsch, A.; Sturm, M.; Reinert, K.; Kohlbacher, O. TOPPAS: A graphical workflow editor for the analysis of high-throughput proteomics data. J. Proteome Res. 2012, 11, 3914–3920. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Scharf, S.S.; Hildebrandt, P.; Burian, M.; Bernhardt, J.; Dhople, V.; Kalinka, J.; Gutjahr, M.; Hammer, E.; Volker, U. Time-resolved quantitative proteome profiling of host-pathogen interactions: The response of Staphylococcus aureus RN1HG to internalisation by human airway epithelial cells. Proteomics 2010, 10, 2801–2811. [Google Scholar] [CrossRef] [PubMed]
- Surmann, K.; Simon, M.; Hildebrandt, P.; Pfortner, H.; Michalik, S.; Stentzel, S.; Steil, L.; Dhople, V.M.; Bernhardt, J.; Schluter, R.; et al. A proteomic perspective of the interplay of Staphylococcus aureus and human alveolar epithelial cells during infection. J. Proteom. 2015, 128, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Corthals, G.L.; Wasinger, V.C.; Hochstrasser, D.F.; Sanchez, J.C. The dynamic range of protein expression: A challenge for proteomic research. Electrophoresis 2000, 21, 1104–1115. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Bakalarski, C.E.; Elias, J.E.; Villen, J.; Haas, W.; Gerber, S.A.; Everley, P.A.; Gygi, S.P. The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J. Proteome Res. 2008, 7, 4756–4765. [Google Scholar] [CrossRef] [PubMed]
- Magdeldin, S.; Moresco, J.J.; Yamamoto, T.; Yates, J.R., III. Off-line multidimensional liquid chromatography and auto sampling result in sample loss in LC/LC-MS/MS. J. Proteome Res. 2014, 13, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Lehmann, T.; Rhode, H. Effect of quality characteristics of single sample preparation steps in the precision and coverage of proteomic studies—A review. Anal. Chim. Acta 2013, 776, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pesek, J.; Krüger, T.; Tautkus, B.; Rhode, H. Colorful quality control of chromatographic sample preparation. J. Chromatogr. B 2013, 934, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Ting, L.; Rad, R.; Gygi, S.P.; Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, N.; Morisaka, H.; Aoki, W.; Takeda, Y.; Shibasaki, S.; Kuroda, K.; Ueda, M. Description of the interaction between Candida albicans and macrophages by mixed and quantitative proteome analysis without isolation. AMB Express 2015, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ju, Z.; Lu, Y.; Mills, G.B.; Akbani, R. A comprehensive comparison of normalization methods for loading control and variance stabilization of reverse-phase protein array data. Cancer Inform. 2014, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger, T.; Luo, T.; Schmidt, H.; Shopova, I.; Kniemeyer, O. Challenges and Strategies for Proteome Analysis of the Interaction of Human Pathogenic Fungi with Host Immune Cells. Proteomes 2015, 3, 467-495. https://doi.org/10.3390/proteomes3040467
Krüger T, Luo T, Schmidt H, Shopova I, Kniemeyer O. Challenges and Strategies for Proteome Analysis of the Interaction of Human Pathogenic Fungi with Host Immune Cells. Proteomes. 2015; 3(4):467-495. https://doi.org/10.3390/proteomes3040467
Chicago/Turabian StyleKrüger, Thomas, Ting Luo, Hella Schmidt, Iordana Shopova, and Olaf Kniemeyer. 2015. "Challenges and Strategies for Proteome Analysis of the Interaction of Human Pathogenic Fungi with Host Immune Cells" Proteomes 3, no. 4: 467-495. https://doi.org/10.3390/proteomes3040467
APA StyleKrüger, T., Luo, T., Schmidt, H., Shopova, I., & Kniemeyer, O. (2015). Challenges and Strategies for Proteome Analysis of the Interaction of Human Pathogenic Fungi with Host Immune Cells. Proteomes, 3(4), 467-495. https://doi.org/10.3390/proteomes3040467