
Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals and Materials
2.2. Animal Model of X-Linked Muscular Dystrophy
2.3. Preparation of Tissue Extracts from Normal and Dystrophic Hind Limb Muscles
2.4. Sample Preparation for Label-Free Liquid Chromatography Mass Spectrometry
2.5. Label-Free Liquid Chromatography Mass Spectrometric Analysis
2.6. Quantitative Proteomic Profiling by Label-Free LC-MS/MS Analysis
2.7. Independent Verification of Key Proteomic Hits by Comparative Immunoblot Analysis
2.8. Immunofluorescence Microscopy
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Protein Name | Unique Peptides | Confidence Score | Anova (p) | Fold Change |
|---|---|---|---|---|---|
| P11531 | Dystrophin Dp427 | 9 | 457.7 | 0.000053 | −14.61 |
| Q8VCT4 | Carboxylesterase 1D | 4 | 175.0 | 0.001943 | −5.85 |
| O55137 | Acyl-coenzyme A thioesterase 1 | 2 | 139.31 | 0.002283 | −4.35 |
| P19096 | Fatty acid synthase | 3 | 198.37 | 0.006235 | −3.99 |
| Q9JJW5 | Myozenin-2 | 2 | 67.3 | 0.016575 | −3.98 |
| Q8C0M9 | Isoaspartyl peptidase/L-asparaginase | 3 | 202.6 | 0.000870 | −3.97 |
| P97823 | Acyl-protein thioesterase 1 | 2 | 115.7 | 0.017276 | −3.96 |
| P32848 | Parvalbumin alpha | 4 | 262.0 | 0.040028 | −3.94 |
| Q61234 | Alpha-1-syntrophin | 3 | 101.6 | 0.000873 | −3.68 |
| P51667 | Myosin regulatory light chain MLC2, slow | 6 | 319.1 | 0.029470 | −3.64 |
| Q08642 | Protein-arginine deiminase type-2 | 10 | 498.3 | 0.003334 | −3.55 |
| P09542 | Myosin light chain MLC3 | 8 | 548.5 | 0.016031 | −3.50 |
| P16015 | Carbonic anhydrase CA3 | 21 | 1652.1 | 0.001537 | −3.43 |
| Q8QZS1 | 3-hydroxyisobutyryl-CoA hydrolase, mitochondrial | 3 | 202.7 | 0.002445 | −3.14 |
| Q64105 | Sepiapterin reductase | 7 | 515.0 | 0.002035 | −3.14 |
| P70695 | Fructose-1,6-bisphosphatase isozyme 2 | 9 | 592.7 | 0.003223 | −3.09 |
| Q9DAK9 | 14 kDa phosphohistidine phosphatase | 2 | 208.6 | 0.000263 | −3.01 |
| P04247 | Myoglobin | 16 | 1768.3 | 0.000652 | −2.93 |
| Q8BVI4 | Dihydropteridine reductase | 5 | 336.0 | 0.000315 | −2.91 |
| Q9DBB8 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | 2 | 119.5 | 0.026280 | −2.90 |
| Q9D358 | Low molecular weight phosphotyrosine protein phosphatase | 2 | 115.6 | 0.002473 | −2.89 |
| P06801 | NADP-dependent malic enzyme | 8 | 348.2 | 0.005720 | −2.84 |
| P21107 | Tropomyosin alpha-3 chain | 3 | 113.6 | 0.024475 | −2.71 |
| O55103 | Periaxin | 2 | 50.9 | 0.014583 | −2.66 |
| Q8R1G2 | Carboxymethylenebutenolidase homolog | 4 | 418.4 | 0.005828 | −2.64 |
| Q9D0K2 | Succinyl-CoA:3-ketoacid coenzyme A transferase 1, mitochondrial | 2 | 143.4 | 0.009917 | −2.60 |
| Q9WUZ5 | Troponin I, slow skeletal muscle | 2 | 114.0 | 0.033414 | −2.50 |
| P56375 | Acylphosphatase-2 | 3 | 227.8 | 0.000551 | −2.39 |
| P17563 | Selenium-binding protein 1 | 7 | 398.9 | 0.000733 | −2.35 |
| P14152 | Malate dehydrogenase, cytoplasmic | 8 | 588.6 | 0.000441 | −2.33 |
| Q9CRB9 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 3, mitochondrial | 2 | 191.9 | 0.008307 | −2.28 |
| P70349 | Histidine triad nucleotide-binding protein 1 | 6 | 504.1 | 0.004703 | −2.27 |
| P11404 | Fatty acid-binding protein, FABP3, heart | 5 | 407.2 | 0.011413 | −2.21 |
| Q01768 | Nucleoside diphosphate kinase B | 3 | 216.1 | 0.029418 | −2.21 |
| Q9D0S9 | Histidine triad nucleotide-binding protein 2, mitochondrial | 2 | 88.7 | 0.007985 | −2.14 |
| Q9CQR4 | Acyl-coenzyme A thioesterase 13 | 2 | 155.3 | 0.009677 | −2.14 |
| P15626 | Glutathione S-transferase Mu 2 | 5 | 300.4 | 0.019473 | −2.13 |
| P08228 | Superoxide dismutase [Cu-Zn] | 5 | 205.9 | 0.004575 | −2.08 |
| Q8BZA9 | Fructose-2,6-bisphosphatase (TIGAR) | 2 | 106.5 | 0.003999 | −2.08 |
| Q91ZJ5 | UTP-glucose-1-phosphate uridylyltransferase | 15 | 949.2 | 0.002697 | −2.08 |
| P63017 | Heat shock cognate 71 kDa | 4 | 465.8 | 0.003124 | −2.03 |
| P15327 | Bisphosphoglycerate mutase | 3 | 213.3 | 0.013366 | −2.02 |
| Q60864 | Stress-induced-phosphoprotein 1 | 3 | 175.8 | 0.001854 | −2.01 |
| Accession No. | Protein Name | Unique Peptides | Confidence Score | Anova (p) | Fold Change |
|---|---|---|---|---|---|
| Q61879 | Myosin-10 | 2 | 89.2 | 0.000004 | mdx only |
| P11276 | Fibronectin | 2 | 133.4 | 0.002667 | 271.95 |
| Q00898 | Alpha-1-antitrypsin 1–5 | 5 | 571.1 | 0.000004 | 187.00 |
| Q02788 | Collagen alpha-2(VI) chain | 2 | 102.6 | 0.000993 | 44.37 |
| P28653 | Biglycan | 4 | 232.4 | 0.004166 | 17.82 |
| Q8R5J9 | PRA1 family protein 3 | 2 | 45.7 | 0.000212 | 16.11 |
| Q9ESD7 | Dysferlin | 2 | 62.5 | 0.004132 | 16.08 |
| Q8VDD5 | Myosin-9 | 4 | 267.7 | 0.009126 | 15.75 |
| Q9D154 | Leukocyte elastase inhibitor A | 9 | 608.0 | 0.000120 | 12.58 |
| P09541 | Myosin light chain MLC4 | 7 | 467.0 | 0.000000 | 12.04 |
| Q8VCM7 | Fibrinogen gamma chain | 3 | 75.5 | 0.000341 | 11.00 |
| Q8K0E8 | Fibrinogen beta chain | 11 | 586.9 | 0.000203 | 10.96 |
| P62835 | Ras-related protein Rap-1A | 2 | 111.6 | 0.004823 | 10.62 |
| P99024 | Tubulin beta-5 chain | 4 | 211.9 | 0.001015 | 9.81 |
| Q3TMP8 | Trimeric intracellular cation channel type A | 2 | 192.7 | 0.004054 | 8.93 |
| P21981 | Protein-glutamine gamma-glutamyltransferase 2 | 2 | 98.6 | 0.023285 | 8.77 |
| O89053 | Coronin-1A | 2 | 83.2 | 0.004520 | 8.09 |
| P51881 | ADP/ATP translocase 2 | 2 | 56.4 | 0.004098 | 8.06 |
| P97449 | Aminopeptidase N | 2 | 60.6 | 0.002562 | 7.79 |
| Q9D1G3 | Protein-cysteine N-palmitoyltransferase HHAT-like protein | 5 | 363.5 | 0.009730 | 6.54 |
| P28665 | Murinoglobulin-1 | 15 | 759.7 | 0.002456 | 6.50 |
| P03921 | NADH-ubiquinone oxidoreductase chain 5 | 3 | 200.0 | 0.018874 | 6.27 |
| Q8VDN2 | Sodium/potassium-transporting ATPase subunit alpha-1 | 5 | 274.2 | 0.015103 | 6.07 |
| P68433 | Histone H3.1 | 3 | 261.4 | 0.011746 | 5.98 |
| P11087 | Collagen alpha-1(I) chain | 4 | 205.2 | 0.016481 | 5.91 |
| Q99JY9 | Actin-related protein 3 | 3 | 152.9 | 0.000084 | 5.58 |
| Q7TSH2 | Phosphorylase b kinase regulatory subunit beta | 2 | 72.7 | 0.035782 | 5.49 |
| Q99MQ4 | Asporin | 5 | 354.1 | 0.000546 | 5.42 |
| Q61233 | Plastin-2 | 6 | 251.2 | 0.000108 | 5.38 |
| P41216 | Long-chain-fatty-acid-CoA ligase 1 | 5 | 242.7 | 0.020046 | 5.19 |
| Q91V79 | Fat storage-inducing transmembrane protein 1 | 2 | 114.1 | 0.012321 | 5.15 |
| Q6PIE5 | Sodium/potassium-transporting ATPase subunit alpha-2 | 6 | 279.1 | 0.010979 | 5.02 |
| P10107 | Annexin A1 | 5 | 349.4 | 0.000751 | 4.98 |
| Q68FD5 | Clathrin heavy chain 1 | 10 | 600.2 | 0.000416 | 4.81 |
| Q00623 | Apolipoprotein A-I | 14 | 959.8 | 0.000386 | 4.63 |
| A2AMM0 | Muscle-related coiled-coil protein | 4 | 173.6 | 0.000173 | 4.60 |
| Q9CR62 | Mitochondrial 2-oxoglutarate/malate carrier protein | 2 | 61.5 | 0.032119 | 4.60 |
| P14094 | Sodium/potassium-transporting ATPase subunit beta-1 | 2 | 99.2 | 0.010749 | 4.38 |
| Q9DBG6 | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 2 | 2 | 123.2 | 0.017838 | 4.26 |
| P13020 | Gelsolin | 10 | 696.4 | 0.000002 | 4.17 |
| P62908 | 40S ribosomal protein S3 | 3 | 191.5 | 0.015578 | 4.15 |
| Q6ZWV3 | 60S ribosomal protein L10 | 2 | 130.3 | 0.035667 | 4.11 |
| Q01339 | Beta-2-glycoprotein 1 | 3 | 76.3 | 0.016181 | 4.11 |
| Q61147 | Ceruloplasmin | 2 | 104.5 | 0.005975 | 4.08 |
| Q60854 | Serpin B6 | 13 | 834.5 | 0.000105 | 3.99 |
| Q99P72 | Reticulon-4 | 2 | 145.7 | 0.007711 | 3.95 |
| Q8BH59 | Calcium-binding mitochondrial carrier protein Aralar1 | 7 | 511.8 | 0.014147 | 3.91 |
| P20918 | Plasminogen | 2 | 160.2 | 0.018437 | 3.88 |
| O09161 | Calsequestrin-2 | 3 | 162.3 | 0.000414 | 3.87 |
| Q61838 | Alpha-2-macroglobulin | 27 | 1386.6 | 0.004622 | 3.78 |
| P16546 | Spectrin alpha chain, non-erythrocytic 1 | 5 | 348.1 | 0.006923 | 3.74 |
| O89104 | Synaptophysin-like protein 2 | 2 | 236.0 | 0.009844 | 3.73 |
| E9PZQ0 | Ryanodine receptor 1 | 27 | 1821.4 | 0.009260 | 3.71 |
| Q71LX4 | Talin-2 | 2 | 46.7 | 0.024488 | 3.69 |
| P68369 | Tubulin alpha-1A chain | 12 | 719.0 | 0.006406 | 3.67 |
| P22752 | Histone H2A type 1 | 4 | 199.0 | 0.001916 | 3.67 |
| P29621 | Serine protease inhibitor A3C | 2 | 240.9 | 0.002558 | 3.66 |
| P01872 | Ig mu chain C region | 3 | 242.2 | 0.004580 | 3.65 |
| Q9EQK5 | Major vault protein | 2 | 108.4 | 0.002870 | 3.58 |
| P07356 | Annexin A2 | 9 | 662.8 | 0.000916 | 3.55 |
| P00405 | Cytochrome c oxidase subunit 2 | 3 | 118.8 | 0.015387 | 3.52 |
| P22599 | Alpha-1-antitrypsin 1-2 | 3 | 186.1 | 0.004003 | 3.46 |
| Q6ZWY9 | Histone H2B type 1-C/E/G | 3 | 310.5 | 0.006482 | 3.46 |
| P26039 | Talin-1 | 3 | 199.2 | 0.001177 | 3.36 |
| P18826 | Phosphorylase b kinase regulatory subunit alpha, skeletal muscle isoform | 3 | 235.7 | 0.008094 | 3.34 |
| P15864 | Histone H1.2 | 2 | 94.9 | 0.011066 | 3.28 |
| P62806 | Histone H4 | 5 | 312.4 | 0.010666 | 3.28 |
| Q8BMS1 | Trifunctional enzyme subunit alpha, mitochondrial | 9 | 762.3 | 0.011513 | 3.25 |
| P26041 | Moesin | 5 | 180.3 | 0.000563 | 3.23 |
| P14148 | 60S ribosomal protein L7 | 3 | 181.6 | 0.013313 | 3.22 |
| Q8VEM8 | Phosphate carrier protein, mitochondrial | 5 | 264.8 | 0.014880 | 3.22 |
| Q61207 | Sulfated glycoprotein 1 | 3 | 122.3 | 0.003193 | 3.13 |
| Q04857 | Collagen alpha-1(VI) chain | 3 | 87.7 | 0.018019 | 3.12 |
| P61027 | Ras-related protein Rab-10 | 3 | 118.3 | 0.001577 | 3.12 |
| P28654 | Decorin | 5 | 350.6 | 0.016544 | 3.10 |
| Q9D783 | Kelch-like protein 40 | 6 | 330.1 | 0.001034 | 3.09 |
| P97927 | Laminin subunit alpha-4 | 3 | 144.0 | 0.002278 | 3.03 |
| O08532 | Voltage-dependent calcium channel subunit alpha-2/delta-1 | 5 | 289.9 | 0.011742 | 3.03 |
| Q8BTM8 | Filamin-A | 3 | 123.0 | 0.006675 | 2.98 |
| A2AUC9 | Kelch-like protein 41 | 12 | 774.9 | 0.008240 | 2.96 |
| P07758 | Alpha-1-antitrypsin 1-1 | 9 | 691.9 | 0.014277 | 2.93 |
| Q9CVB6 | Actin-related protein 2/3 complex subunit 2 | 3 | 154.1 | 0.000307 | 2.91 |
| P18760 | Cofilin-1 | 5 | 305.0 | 0.004511 | 2.90 |
| Q8CI43 | Myosin light chain 6B | 2 | 66.6 | 0.007575 | 2.89 |
| P48962 | ADP/ATP translocase 1 | 9 | 579.2 | 0.017242 | 2.85 |
| Q9D6F9 | Tubulin beta-4A chain | 5 | 478.9 | 0.003730 | 2.78 |
| Q8R429 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | 39 | 3489.2 | 0.019926 | 2.77 |
| P09405 | Nucleolin | 2 | 453.6 | 0.005112 | 2.75 |
| P24369 | Peptidyl-prolyl cis-trans isomerase B | 2 | 132.9 | 0.000596 | 2.72 |
| P13541 | Myosin-3 | 5 | 425.1 | 0.018031 | 2.71 |
| P48036 | Annexin A5 | 8 | 459.7 | 0.003110 | 2.70 |
| P29391 | Ferritin light chain 1 | 9 | 652.0 | 0.005573 | 2.68 |
| P62874 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 | 3 | 178.5 | 0.001344 | 2.67 |
| P32261 | Antithrombin-III | 3 | 178.1 | 0.004703 | 2.65 |
| P07759 | Serine protease inhibitor A3K | 9 | 716.8 | 0.002934 | 2.64 |
| Q7TMM9 | Tubulin beta-2A chain | 10 | 632.2 | 0.001380 | 2.64 |
| P47911 | 60S ribosomal protein L6 | 2 | 145.7 | 0.011238 | 2.59 |
| Q91X72 | Hemopexin | 13 | 636.3 | 0.016945 | 2.58 |
| P14869 | 60S acidic ribosomal protein | 3 | 86.0 | 0.004999 | 2.56 |
| Q9JK53 | Prolargin | 5 | 227.2 | 0.005281 | 2.56 |
| P48678 | Prelamin-A/C | 25 | 1641.1 | 0.000349 | 2.55 |
| Q9CQQ7 | ATP synthase F(0) complex subunit B1, mitochondrial | 5 | 211.7 | 0.003672 | 2.54 |
| Q9CZM2 | 60S ribosomal protein L15 | 2 | 104.9 | 0.024828 | 2.51 |
| P62962 | Profilin-1 | 4 | 270.9 | 0.015520 | 2.49 |
| Q91VI7 | Ribonuclease inhibitor | 7 | 339.1 | 0.013370 | 2.48 |
| Q07076 | Annexin A7 | 2 | 120.8 | 0.001600 | 2.46 |
| Q8R4E4 | Myozenin-3 | 2 | 100.5 | 0.001180 | 2.45 |
| P62141 | Serine/threonine-protein phosphatase PP1-beta catalytic subunit | 2 | 177.1 | 0.001508 | 2.45 |
| Q70IV5 | Synemin | 2 | 104.4 | 0.000186 | 2.43 |
| P20152 | Vimentin | 15 | 1088.5 | 0.002258 | 2.42 |
| P35980 | 60S ribosomal protein L18 | 3 | 154.7 | 0.018170 | 2.41 |
| Q62000 | Mimecan | 7 | 497.7 | 0.016065 | 2.41 |
| Q60930 | Voltage-dependent anion-selective channel protein 2 | 9 | 649.6 | 0.040913 | 2.36 |
| Q8BFR5 | Elongation factor Tu, mitochondrial | 9 | 509.1 | 0.004815 | 2.36 |
| Q9WTR5 | Cadherin-13 | 3 | 245.5 | 0.006036 | 2.36 |
| P47757 | F-actin-capping protein subunit beta | 2 | 143.1 | 0.003431 | 2.34 |
| Q9Z1E4 | Glycogen [starch] synthase, muscle | 4 | 199.6 | 0.019418 | 2.32 |
| P97384 | Annexin A11 | 5 | 201.6 | 0.000422 | 2.32 |
| P68040 | Guanine nucleotide-binding protein subunit beta-2-like 1 | 6 | 311.8 | 0.000693 | 2.31 |
| Q91WD5 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial | 8 | 305.4 | 0.001414 | 2.31 |
| P47963 | 60S ribosomal protein L13 | 2 | 109.2 | 0.023095 | 2.30 |
| P50543 | Protein S100-A11 | 2 | 136.8 | 0.003349 | 2.30 |
| Q61425 | Hydroxyacyl-coenzyme A dehydrogenase, mitochondrial | 2 | 115.4 | 0.012822 | 2.29 |
| P05213 | Tubulin alpha-1B chain | 2 | 209.9 | 0.016043 | 2.29 |
| P68368 | Tubulin alpha-4A chain | 3 | 305.2 | 0.006357 | 2.28 |
| Q8BG05 | Heterogeneous nuclear ribonucleoprotein A3 | 2 | 82.4 | 0.040172 | 2.28 |
| Q6ZWX6 | Eukaryotic translation initiation factor 2 subunit 1 | 2 | 73.2 | 0.018602 | 2.27 |
| A2AAJ9 | Obscurin | 9 | 474.2 | 0.004426 | 2.26 |
| P62889 | 60S ribosomal protein L30 | 4 | 279.4 | 0.001887 | 2.26 |
| Q9DC69 | NADH dehydrogenase 1 alpha subcomplex subunit 9, mitochondrial | 3 | 184.7 | 0.014362 | 2.26 |
| P50544 | Very long-chain specific acyl-CoA dehydrogenase, mitochondrial | 3 | 148.7 | 0.002723 | 2.25 |
| P63101 | 14-3-3 protein zeta/delta | 4 | 390.8 | 0.006991 | 2.23 |
| Q8BK84 | Dual specificity phosphatase DUPD1 | 2 | 146.8 | 0.004756 | 2.21 |
| Q9DB20 | ATP synthase subunit O, mitochondrial | 4 | 277.8 | 0.010221 | 2.18 |
| Q3MI48 | Junctional sarcoplasmic reticulum protein 1 | 3 | 145.6 | 0.020961 | 2.17 |
| P14824 | Annexin A6 | 5 | 539.1 | 0.003047 | 2.15 |
| Q03265 | ATP synthase subunit alpha, mitochondrial | 11 | 778.3 | 0.001158 | 2.10 |
| Q9JJZ2 | Tubulin alpha-8 chain | 2 | 68.2 | 0.023642 | 2.10 |
| Q60605 | Myosin light polypeptide 6 | 4 | 300.2 | 0.010440 | 2.09 |
| Q99JB8 | Protein kinase C and casein kinase II substrate protein 3 | 8 | 439.7 | 1.20E-05 | 2.09 |
| P31001 | Desmin | 18 | 1622.8 | 0.001451 | 2.09 |
| Q61292 | Laminin subunit beta-2 | 5 | 233.6 | 0.013369 | 2.08 |
| P40124 | Adenylyl cyclase-associated protein 1 | 2 | 52.1 | 0.001465 | 2.07 |
| Q8VHX6 | Filamin-C | 20 | 1257.4 | 0.038298 | 2.07 |
| P51885 | Lumican | 5 | 346.1 | 0.021066 | 2.07 |
| Q9CZU6 | Citrate synthase, mitochondrial | 5 | 373.2 | 0.014760 | 2.07 |
| O35129 | Prohibitin-2 | 3 | 122.3 | 0.004054 | 2.06 |
| Q9DB60 | Prostamide/prostaglandin F synthase | 2 | 48.5 | 0.001144 | 2.05 |
| O88342 | WD repeat-containing protein 1 | 3 | 205.6 | 0.004625 | 2.04 |
| P14602 | Heat shock protein beta-1 | 6 | 349.6 | 0.002016 | 2.04 |
| P14733 | Lamin-B1 | 5 | 244.4 | 0.001897 | 2.03 |
| Q60936 | Chaperone activity of bc1 complex-like, mitochondrial | 2 | 98.4 | 0.028276 | 2.02 |
| P35979 | 60S ribosomal protein L12 | 3 | 172.6 | 0.020128 | 2.02 |
| Q02053 | Ubiquitin-like modifier-activating enzyme 1 | 2 | 170.9 | 0.030810 | 2.01 |
3.1. Label-Free LC-MS/MS Analysis of Decreased Proteins in Total mdx-4cv Muscle Extracts
3.2. Label-Free LC-MS/MS Analysis of Increased Proteins in Total mdx-4cv Muscle Extracts
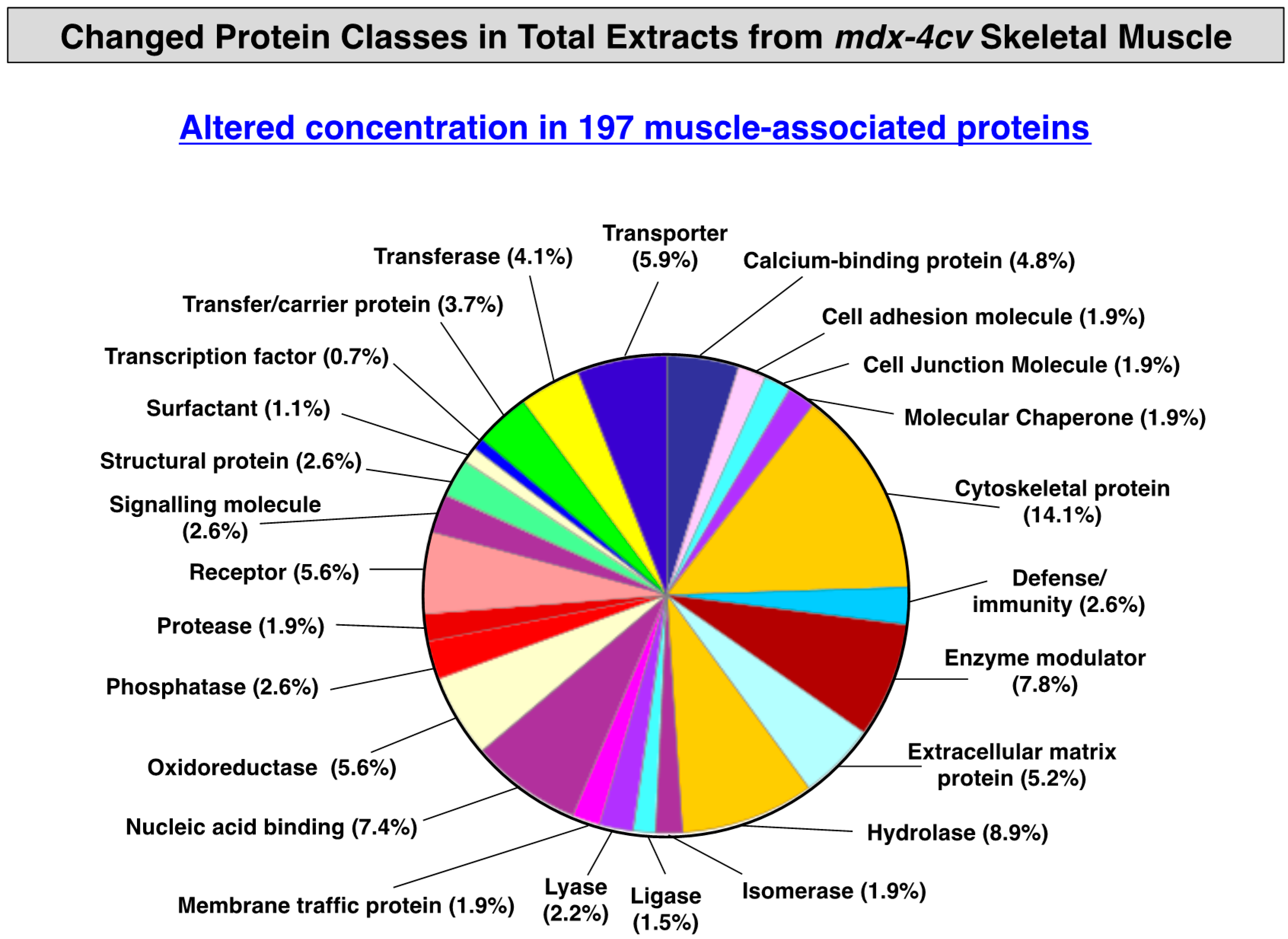
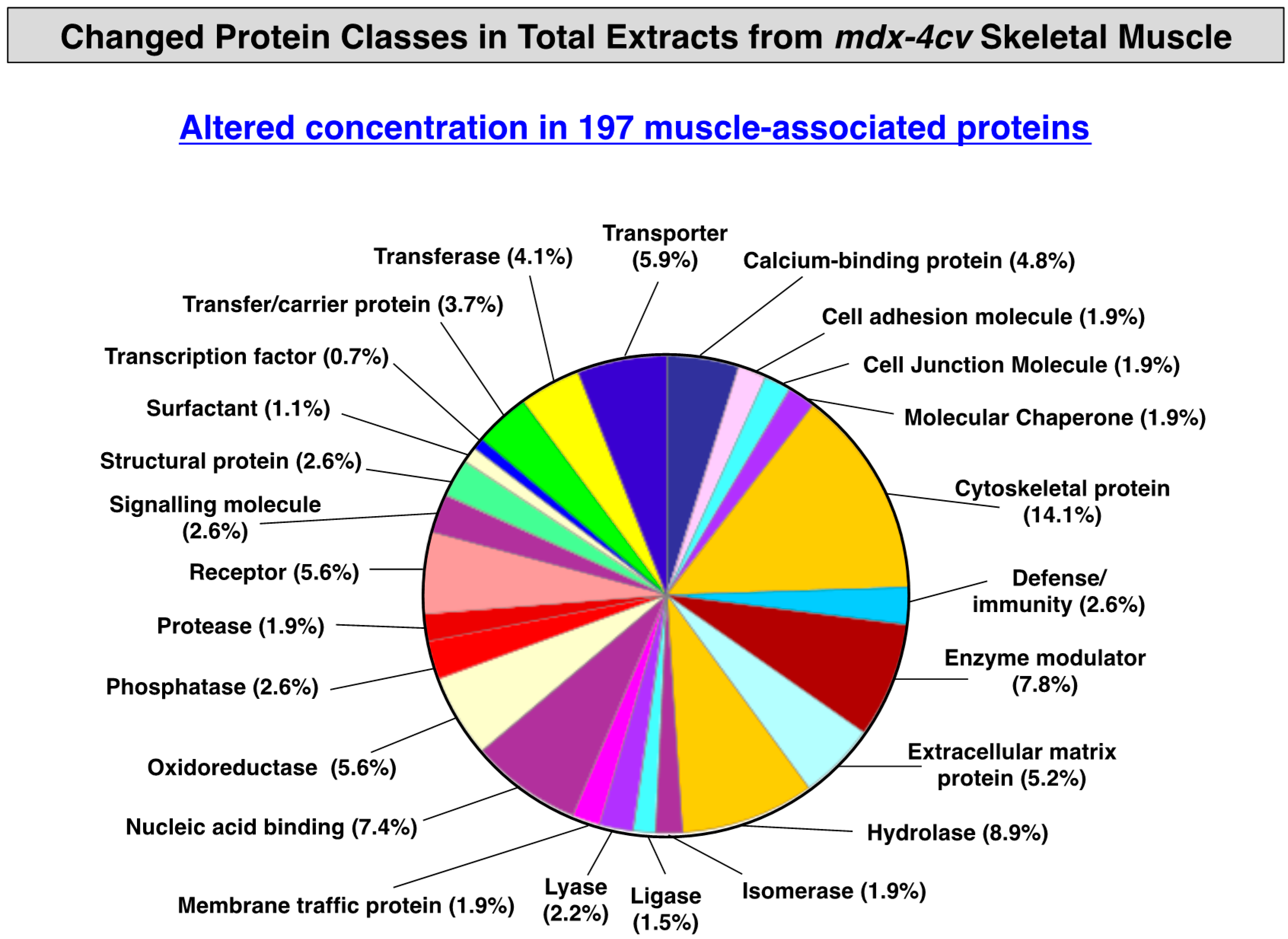
3.3. Distribution of Protein Changes in Dystrophic mdx-4cv Hind Limb Muscles
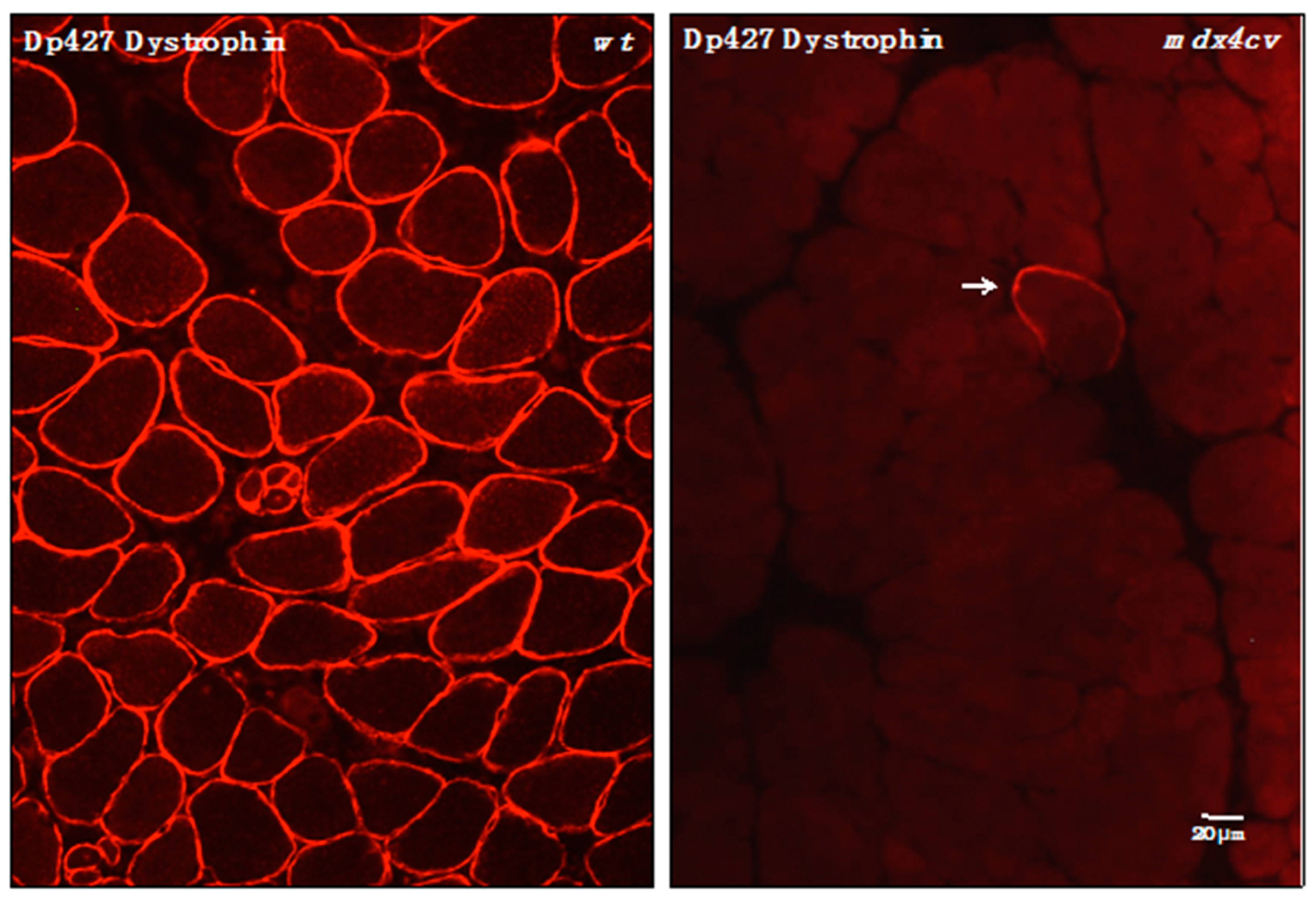
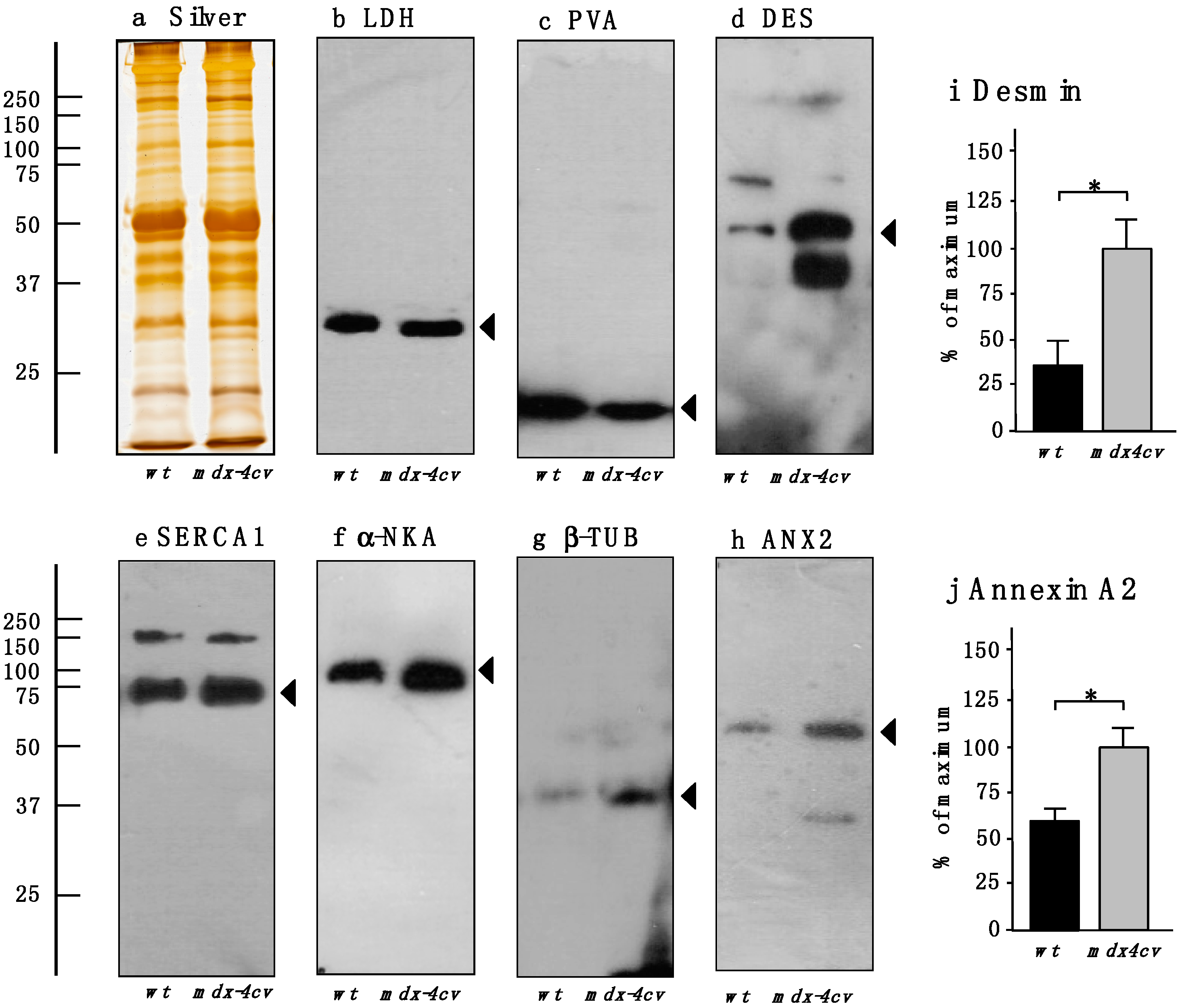
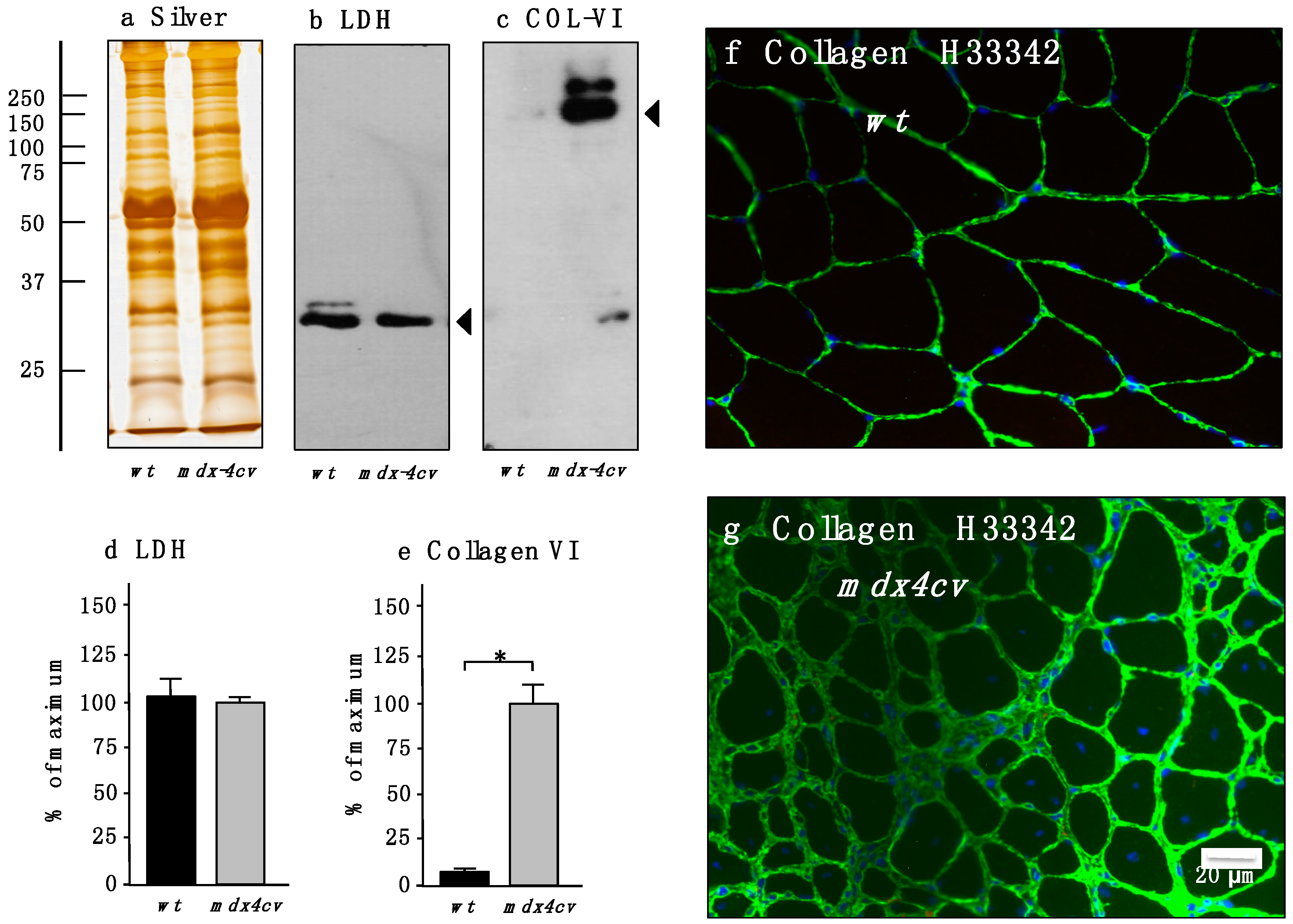
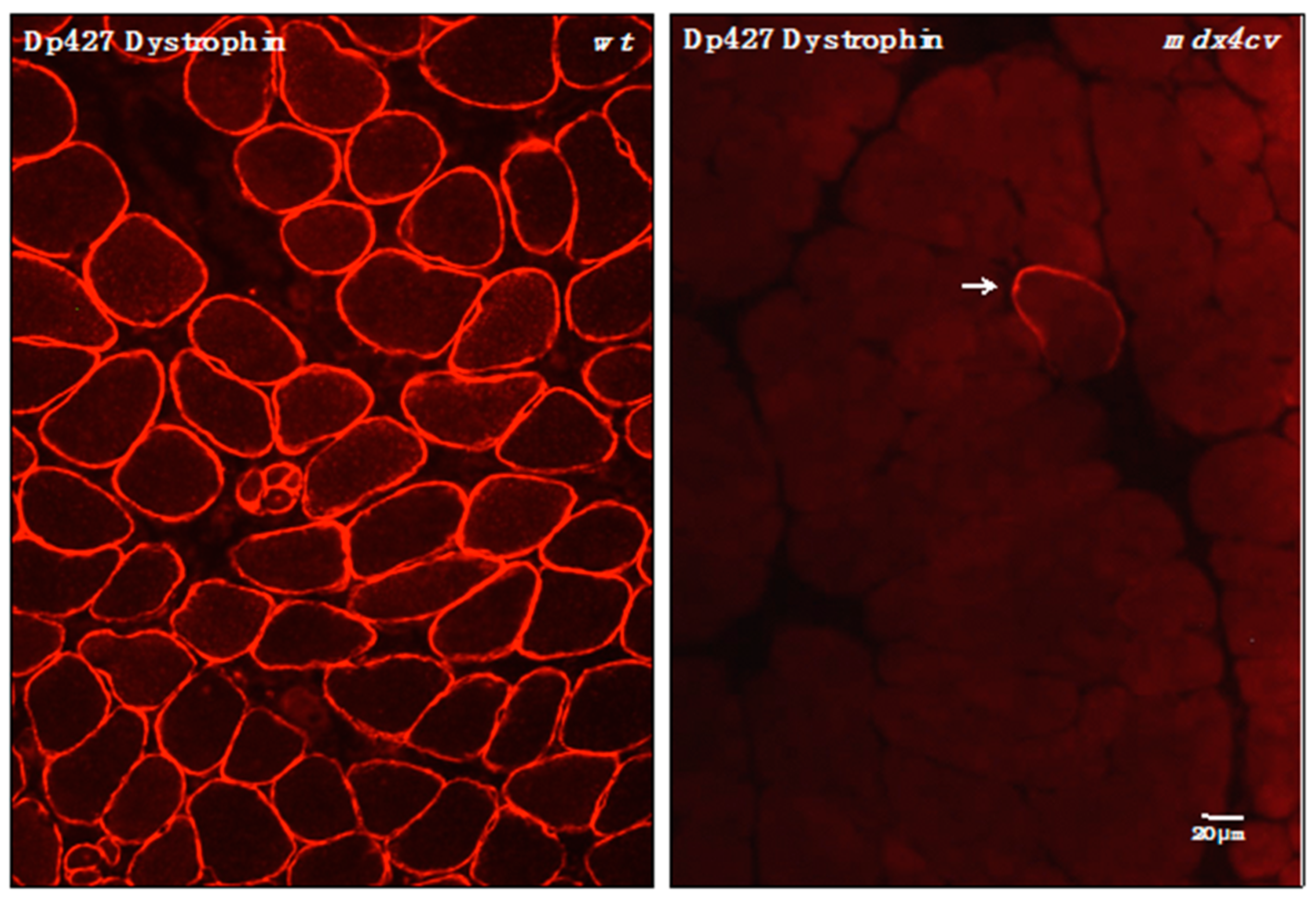
3.4. Verification of Proteomic Changes in Dystrophic mdx-4cv Hind Limb Muscles


4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [PubMed]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.M.; Lin, A.Y.; Thomas, D.D.; Ervasti, J.M. The carboxy-terminal third of dystrophin enhances actin binding activity. J. Mol. Biol. 2012, 416, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystro- phin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Beggs, A.H.; Hoffman, E.P.; Snyder, J.R.; Arahata, K.; Specht, L.; Shapiro, F.; Angelini, C.; Sugita, H.; Kunkel, L.M. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: Dystrophin gene and protein studies. Am. J. Hum. Genet. 1991, 49, 54–67. [Google Scholar] [PubMed]
- Diegoli, M.; Grasso, M.; Favalli, V.; Serio, A.; Gambarin, F.I.; Klersy, C.; Pasotti, M.; Agozzino, E.; Scelsi, L.; Ferlini, A.; et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J. Am. Coll. Cardiol. 2011, 58, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomics of skeletal muscle differentiation, neuromuscular disorders and fiber aging. Expert Rev. Proteomics 2010, 7, 283–296. [Google Scholar] [PubMed]
- Burniston, J.G.; Hoffman, E.P. Proteomic responses of skeletal and cardiac muscle to exercise. Expert Rev. Proteomics 2011, 8, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of human skeletal muscle in health and disease: Contribution of proteomics. J. Proteomics 2011, 74, 774–795. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Holland, A.; Ohlendieck, K. Mass spectrometry-based identification of muscle-associated and muscle-derived proteomic biomarkers of dystrophinopathies. J. Neuromuscul. Dis. 2014, 1, 15–40. [Google Scholar]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Proteomic analysis of mdx skeletal muscle: Great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 2003, 3, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Dowling, P.; Lohan, J.; McDonnell, K.; Poetsch, S.; Ohlendieck, K. Subproteomics analysis of Ca2+-binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. Eur. J. Biochem. 2004, 271, 3943–3952. [Google Scholar] [PubMed]
- Doran, P.; Dowling, P.; Donoghue, P.; Buffini, M.; Ohlendieck, K. Reduced expression of regucalcin in young and aged mdx diaphragm indicates abnormal cytosolic calcium handling in dystrophin-deficient muscle. Biochim. Biophys. Acta 2006, 1764, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Brinkmeier, H.; Zhang, Y.; Winkler, C.K.; Ohlendieck, K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscle from the mdx mouse model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 544–556. [Google Scholar] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Profiling of age-related changes in the tibialis anterior muscle proteome of the mdx mouse model of dystrophinopathy. J. Biomed. Biotechnol. 2012, 2012, 691641. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Wilton, S.D.; Fletcher, S.; Ohlendieck, K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics 2009, 9, 671–685. [Google Scholar] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative proteomic analysis of the contractile protein-depleted fraction from normal versus dystrophic skeletal muscle. Anal. Biochem. 2014, 446, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeier, H.; Ohlendieck, K. Chaperoning heat shock proteins: Proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteomics Clin. Appl. 2014, 8, 875–895. [Google Scholar] [CrossRef] [PubMed]
- Gardan-Salmon, D.; Dixon, J.M.; Lonergan, S.M.; Selsby, J.T. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur. J. Appl. Physiol. 2011, 111, 2763–2773. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, C.Y.; Menezes de Oliveira, B.; Durbeej, M.; Marques, M.J. Isobaric Tagging-Based Quantification for Proteomic Analysis: A Comparative Study of Spared and Affected Muscles from mdx Mice at the Early Phase of Dystrophy. PLoS ONE 2013, 8, e65831. [Google Scholar] [CrossRef] [PubMed]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in aged mdx diaphragm muscle. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Application of fluorescence two-dimensional difference in-gel electrophoresis as a proteomic biomarker discovery tool in muscular dystrophy research. Biology 2013, 2, 1438–1464. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, V.D.; van Putten, M.; Chaouch, A.; Garrood, P.; Straub, V.; Lochmüller, H.; Ginjaar, H.B.; Aartsma-Rus, A.M.; van Ommen, G.J.; den Dunnen, J.T.; et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul. Disord. 2011, 21, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Duguez, S.; Duddy, W.; Johnston, H.; Lainé, J.; le Bihan, M.C.; Brown, K.J.; Bigot, A.; Hathout, Y.; Butler-Browne, G.; Partridge, T. Dystrophin deficiency leads to disturbance of LAMP1-vesicle-associated protein secretion. Cell. Mol. Life Sci. 2013, 70, 2159–2174. [Google Scholar] [PubMed]
- Martin, C.F.; Hiller, M.; Spitali, P.; Oonk, S.; Dalebout, H.; Palmblad, M.; Chaouch, A.; Guglieri, M.; Straub, V.; Lochmüller, H.; et al. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics Clin. Appl. 2014, 8, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Marathi, R.L.; Rayavarapu, S.; Zhang, A.; Brown, K.J.; Seol, H.; Gordish-Dressman, H.; Cirak, S.; Bello, L.; Nagaraju, K.; et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 2014, 23, 6458–69. [Google Scholar] [PubMed]
- Ayoglu, B.; Chaouch, A.; Lochmüller, H.; Politano, L.; Bertini, E.; Spitali, P.; Hiller, M.; Niks, E.H.; Gualandi, F.; Pontén, F.; et al. Affinity proteomics within rare diseases: A BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014, 6, 918–936. [Google Scholar] [CrossRef] [PubMed]
- Rouillon, J.; Zocevic, A.; Leger, T.; Garcia, C.; Camadro, J.M.; Udd, B.; Wong, B.; Servais, L.; Voit, T.; Svinartchouk, F. Proteomics profiling of urine reveals specific titin fragments as biomarkers of Duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Johnson, E.; Xu, R.; Martin, L.T.; Martin, P.T.; Montanaro, F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 2012, 11, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Ohlendieck, K. Mass spectrometric identification of dystrophin isoform Dp427 by on-membrane digestion of sarcolemma from skeletal muscle. Anal. Biochem. 2010, 404, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Swiderski, K.; Shaffer, S.A.; Gallis, B.; Odom, G.L.; Arnett, A.L.; Scott Edgar, J.; Baum, D.M.; Chee, A.; Naim, T.; Gregorevic, P.; et al. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum. Mol. Genet. 2014, 23, 6697–6711. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.J.; Marathi, R.; Fiorillo, A.A.; Ciccimaro, E.F.; Sharma, S.; Rowlands, D.S.; Rayavarapu, S.; Nagaraju, K.; Hoffman, E.P.; Hathout, Y. Accurate Quantitation of Dystrophin Protein in Human Skeletal Muscle Using Mass Spectrometry. J. Bioanal. Biomed. 2012, Suppl. 7, 001. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous pathoproteomic evaluation of the dystrophin-glycoprotein complex and secondary changes in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biology 2015, 4, 397–423. [Google Scholar] [CrossRef] [PubMed]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model Mech. 2015, 8, 195–213. [Google Scholar] [PubMed]
- Judge, L.M.; Haraguchiln, M.; Chamberlain, J.S. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006, 119, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Ohlendieck, K. Mass spectrometric characterization of the sarcoplasmic reticulum from rabbit skeletal muscle by on-membrane digestion. Protein Pept. Lett. 2012, 19, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [PubMed]
- Meleady, P.; Gallagher, M.; Clarke, C.; Henry, M.; Sanchez, N.; Barron, N.; Clynes, M. Impact of miR-7 over-expression on the proteome of Chinese hamster ovary cells. J. Biotechnol. 2012, 160, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Meleady, P.; Hoffrogge, R.; Henry, M.; Rupp, O.; Bort, J.H.; Clarke, C.; Brinkrolf, K.; Kelly, S.; Müller, B.; Doolan, P.; et al. Utilization and evaluation of CHO-specific sequence databases for mass spectrometry based proteomics. Biotechnol. Bioeng. 2012, 109, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Linge, A.; Maurya, P.; Friedrich, K.; Baretton, G.B.; Kelly, S.; Henry, M.; Clynes, M.; Larkin, A.; Meleady, P. Identification and functional validation of RAD23B as a potential protein in human breast cancer progression. J. Proteome Res. 2014, 13, 3212–3222. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Schmitt-John, T.; Dowling, P.; Meleady, P.; Henry, M.; Clynes, M.; Ohlendieck, K. Intricate effects of primary motor neuronopathy on contractile proteins and metabolic muscle enzymes as revealed by label-free mass spectrometry. Biosci. Rep. 2014, 34, pii: e00119. [Google Scholar] [CrossRef] [PubMed]
- PANTHER Gene List Analysis. Available online: http://pantherdb.org/ (accessed on 4 May 2015).
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- STRING Functional Protein Association Network. Available online: http://string-db.org/ (accessed on 3 February 2015).
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Mundegar, R.R.; Franke, E.; Schäfer, R.; Zweyer, M.; Wernig, A. Reduction of high background staining by heating unfixed mouse skeletal muscle tissue sections allows for detection of thermostable antigens with murine monoclonal antibodies. J. Histochem. Cytochem. 2008, 56, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genomics Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [PubMed]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle 2015, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Murphy, S.; Dowling, D.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy-associated myofibrosis. Proteomics 2015. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. BioMed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, K.; Fadic, R.; Brandan, E. Augmented synthesis and differential localization of heparan sulfate proteoglycans in Duchenne muscular dystrophy. J. Cell. Biochem. 2002, 85, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, S.; Cuellar, C.; Casar, J.C.; Garrido, J.; Schaefer, L.; Kresse, H.; Brandan, E. Synthesis of proteoglycans is augmented in dystrophic mdx mouse skeletal muscle. Eur. J. Cell Biol. 2000, 79, 173–181. [Google Scholar] [CrossRef]
- Zanotti, S.; Negri, T.; Cappelletti, C.; Bernasconi, P.; Canioni, E.; di Blasi, C.; Pegoraro, E.; Angelini, C.; Ciscato, P.; Prelle, A.; et al. Decorin and biglycan expression is differentially altered in several muscular dystrophies. Brain 2005, 128, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Fadic, R.; Mezzano, V.; Alvarez, K.; Cabrera, D.; Holmgren, J.; Brandan, E. Increase in decorin and biglycan in Duchenne Muscular Dystrophy: Role of fibroblasts as cell source of these proteoglycans in the disease. J. Cell. Mol. Med. 2006, 10, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef]
- Zanotti, S.; Saredi, S.; Ruggieri, A.; Fabbri, M.; Blasevich, F.; Romaggi, S.; Morandi, L.; Mora, M. Altered extracellular matrix transcript expression and protein modulation in primary Duchenne muscular dystrophy myotubes. Matrix Biol. 2007, 26, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Gibertini, S.; Mora, M. Altered production of extra-cellular matrix components by muscle-derived Duchenne muscular dystrophy fibroblasts before and after TGF-beta1 treatment. Cell Tissue Res. 2010, 339, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Marshall, P.A.; Williams, P.E.; Goldspink, G. Accumulation of collagen and altered fiber-type ratios as indicators of abnormal muscle gene expression in the mdx dystrophic mouse. Muscle Nerve 1989, 12, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Goldspink, G.; Fernandes, K.; Williams, P.E.; Wells, D.J. Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscul. Disord. 1994, 4, 183–191. [Google Scholar] [CrossRef]
- Trensz, F.; Haroun, S.; Cloutier, A.; Richter, M.V.; Grenier, G. A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am. J. Physiol. Cell Physiol. 2010, 299, C939–C947. [Google Scholar] [CrossRef] [PubMed]
- Lessa, T.B.; de Abreu, D.K.; Rodrigues, M.N.; Brólio, M.P.; Miglino, M.A.; Ambrósio, C.E. Morphological and ultrastructural evaluation of the golden retriever muscular dystrophy trachea, lungs, and diaphragm muscle. Microsc. Res. Tech. 2014, 77, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Comparative Proteomics for Studying Muscular Dystrophy: Intrinsic Biological and Analytical Issues Associated with the Systematic Utilization of Tissue Specimens. J. Proteomics Bioinform. 2014, S10. in press. [Google Scholar] [CrossRef]
- Gelfi, C.; Viganò, A.; de Palma, S.; Ripamonti, M.; Begum, S.; Cerretelli, P.; Wait, R. 2-D protein maps of rat gastrocnemius and soleus muscles: A tool for muscle plasticity assessment. Proteomics 2006, 6, 321–340. [Google Scholar] [CrossRef]
- Drexler, H.C.; Ruhs, A.; Konzer, A.; Mendler, L.; Bruckskotten, M.; Looso, M.; Günther, S.; Boettger, T.; Krüger, M.; Braun, T. On marathons and Sprints: An integrated quantitative proteomics and transcriptomics analysis of differences between slow and fast muscle fibers. Mol. Cell. Proteomics 2012, 11, M111.010801. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G.; Connolly, J.; Kainulainen, H.; Britton, S.L.; Koch, L.G. Label-free profiling of skeletal muscle using high-definition mass spectrometry. Proteomics 2014, 14, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S.; Murgia, M.; Nagaraja, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways and transcription factors. Mol. Cell. Proteomics 2015, 14, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.; Doran, P.; Wynne, K.; Pedersen, K.; Dunn, M.J.; Ohlendieck, K. Proteomic profiling of chronic low-frequency stimulated fast muscle. Proteomics 2007, 7, 3417–3430. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.A.; Cobley, J.N.; Morton, J.P.; Close, G.L.; Edwards, B.J.; Koch, L.G.; Britton, S.L.; Burniston, J.G. Label-Free LC-MS Profiling of Skeletal Muscle Reveals Heart-Type Fatty Acid Binding Protein as a Candidate Biomarker of Aerobic Capacity. Proteomes 2013, 1, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the contractile apparatus from skeletal muscle. Expert Rev. Proteomics 2013, 10, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Henry, M.; Meleady, P.; Winkler, C.K.; Krautwald, M.; Brinkmeier, H.; Ohlendieck, K. Comparative label-free mass spectrometric analysis of mildly versus severely affected mdx mouse skeletal muscles identifies annexin, lamin and vimentin as universal dystrophic markers. Molecules 2015, 20, 11317–11344. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Fritz, D.; Stefanovic, L.; Stefanovic, B. Nonmuscle myosin-dependent synthesis of type I collagen. J. Mol. Biol. 2010, 401, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Lieber, R.L.; Ward, S.R. Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol. 2013, 305, C241–C252. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; de Mori, R.; Baeza-Raja, B.; Martínez de Lagrán, M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, V.; Petrella, A.; Parente, L. Annexin A1: Novel roles in skeletal muscle biology. J. Cell. Physiol. 2012, 227, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Vontzalidis, A.; Terzis, G.; Manta, P. Increased dysferlin expression in Duchenne muscular dystrophy. Anal. Quant. Cytopathol. Histpathol. 2014, 36, 15–22. [Google Scholar] [PubMed]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Müller, M.; Izykowski, N.; Hong, J.; Hong, K.; Kim, S.H.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.C.; Wright, N.T. Structure of giant muscle proteins. Front. Physiol. 2013, 4, 368. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.E.; Froemming, G.R.; Maguire, P.B.; Ohlendieck, K. Excitation-contraction-relaxation cycle: Role of Ca2+-regulatory membrane proteins in normal, stimulated and pathological skeletal muscle (review). Int. J. Mol. Med. 1998, 1, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.F.; Burton, K.A.; Dauncey, M.J. Ouabain sensitive Na+/K+-ATPase content is elevated in mdx mice: Implications for the regulation of ions in dystrophic muscle. J. Neurol. Sci. 1995, 133, 11–15. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, S.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes 2015, 3, 298-327. https://doi.org/10.3390/proteomes3030298
Murphy S, Zweyer M, Mundegar RR, Henry M, Meleady P, Swandulla D, Ohlendieck K. Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes. 2015; 3(3):298-327. https://doi.org/10.3390/proteomes3030298
Chicago/Turabian StyleMurphy, Sandra, Margit Zweyer, Rustam R. Mundegar, Michael Henry, Paula Meleady, Dieter Swandulla, and Kay Ohlendieck. 2015. "Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles" Proteomes 3, no. 3: 298-327. https://doi.org/10.3390/proteomes3030298
APA StyleMurphy, S., Zweyer, M., Mundegar, R. R., Henry, M., Meleady, P., Swandulla, D., & Ohlendieck, K. (2015). Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes, 3(3), 298-327. https://doi.org/10.3390/proteomes3030298