
Activation of SsoPK4, an Archaeal eIF2α Kinase Homolog, by Oxidized CoA

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5ʹ to 3ʹ) | Purpose |
|---|---|---|
| SsoPK4/sso3182 | ||
| rSsoPK4 forward | AGTAATACCATGGCGATTAATCTGTTACAATAC | NcoI at M1 |
| rSsoPK4 reverse | CAGTATGTCGACTCTAAAGTAGAAAAACTCCTTTAT | SalI |
| rSsoPK4(198–635) | TTACTAACCATGGTTCCGAGCAAAGGAATACCTAG | NcoI at L197M |
| rSsoPK4(284–635) | CACCAGCCATGGTTCCGAGCAAAGGAATACCTAG | NcoI at M284 |
| rSsoPK4(320–635) | ATTTTAGCCATGGCGAACTGGGATCCTAAAGTATGGGTAGG | NcoI at M320 |
| rSsoPK4seq.fwd. | CCGAGCAAAGGAATACCTAG | sequencing |
| rSsoPK4 seq. rev. | ACATCGTTTATAGATCCACC | sequencing |
| D476A forward | GAGGGATATGTTCACTGTGCTGTAAAACCTCAAAATG | D476A |
| D476A reverse | CATTTTGAGGTTTTACAGCACAGTGAACATATCCCTC | D476A |
| K363A forward | GGGAATTTTTATGCTCTCGCGATACCGTTAATAAATTAC | K363A |
| K363A reverse | GTAATTTATTAACGGTATCGCGAGAGCATAAAAATTCCC | K363A |
| T606A forward | GAAAATACGTGGATAAAAATGCTTATCTCTTCATATCAAAAATGG | T606A |
| T606A reverse | CCATTTTTGATATGAAGAGATAAGCATTTTTATCCACGTATTTTC | T606A |
| S611A forward | GGATAAAAATACTTATCTCTTCATAGCAAAAATGGTAGATCCGG | S611A |
| S611A reverse | CCGGATCTACCATTTTTGCTATGAAGAGATAAGTATTTTTATCC | S611A |
| T606D forward | GAAAATACGTGGATAAAAATGATTATCTCTTCATATCAAAAATGG | T606D |
| T606D reverse | CCATTTTTGATATGAAGAGATAATCATTTTTTATCCACGTATTTTC | T606D |
| S611D forward | GGATAAAAATACTTATCTCTTCATAGACAAAATGGTAGATCCGG | S611D |
| S611D reverse | CCGGATCTACCATTTTGTCTATGAAGAGATAAGTATTTTTATCC | S611D |
| KKA forward | GCGTTATTATCGAGCAGAAACATAGAGCTATTAGAATTAGC | K295N/K296I |
| KKA reverse | GCTAATTCTAATAGCTCTATGTTTCTGCTCGATAATAACGC | K295N/K296I |
| KKB forward | CTATTAGAATTAGCATGTATAAACGGGTATAAGAAAGCTTG | K304I/K305N |
| KKB reverse | CAAGCTTTCTTATACCCGTTTATACATGCTAATTCTAATAG | K304I/K305N |
| KKC forward | CATGTAAAAAGGGGTATAACATAGCTTGTGAGCAGACTAAAC | K308N/K309I |
| KKC reverse | GTTTAGTCTGCTCACAAGCTATGTTATACCCCTTTTTACATG | K308N/K309I |
| S-tagT2A forward | GCCCAGATCTGGGTGCGCTGGTGCCAGCGG | T2A in S-tag |
| S-tagT2A reverse | CCGCGTGGCACCAGCGCACCAGATCTGGGC | T2A in S-tag |
| aIF2α/sso1050 | ||
| aIF2α forward | GGGTTACCATGGTTTACAGTAGAAGCAAACTACCCTCAG | NcoI at M1 |
| aIF2α reverse | CCTCATTTTCCGTCGACTTTCTTAACCACACTTATATCTACG | SalI |
| S262A forward | GAAGAAAACGTAGATATAGCTGTGGTTAAGAAAGTCGACAAG | S262A |
| S262A reverse | CTTGTCGACTTTCTTAACCACAGCTATATCTACGTTTTCTTC | S262A |
| S47A&S48A forward | GCCTTGGAGTGAAGTAACTACCAAATGGGTTAAGAATATAAGGG | S47A&S48A |
| S47A&S48A reverse | CCCTTATATTCTTAACCCATTTGGTAGTTACTTCACTCCAAGGC | S47A&S48A |
| S47T&S48T forward | GCCTTGGAGTGAAGTAAcTAcCAAATGGGTTAAGAATATAAGGG | S47T&S48T |
| S47T&S48T | CCCTTATATTCTTAACCCATTTGGTAGTTACTTCACTCCAAGGC | S47T&S48T |
2.2. Standard Procedures
2.3. Cloning and Mutagenesis
2.4. Expression and Purification of Recombinant Proteins
2.5. Assay of Protein Kinase Activity
2.6. Sucrose Density Gradient Ultracentrifugation
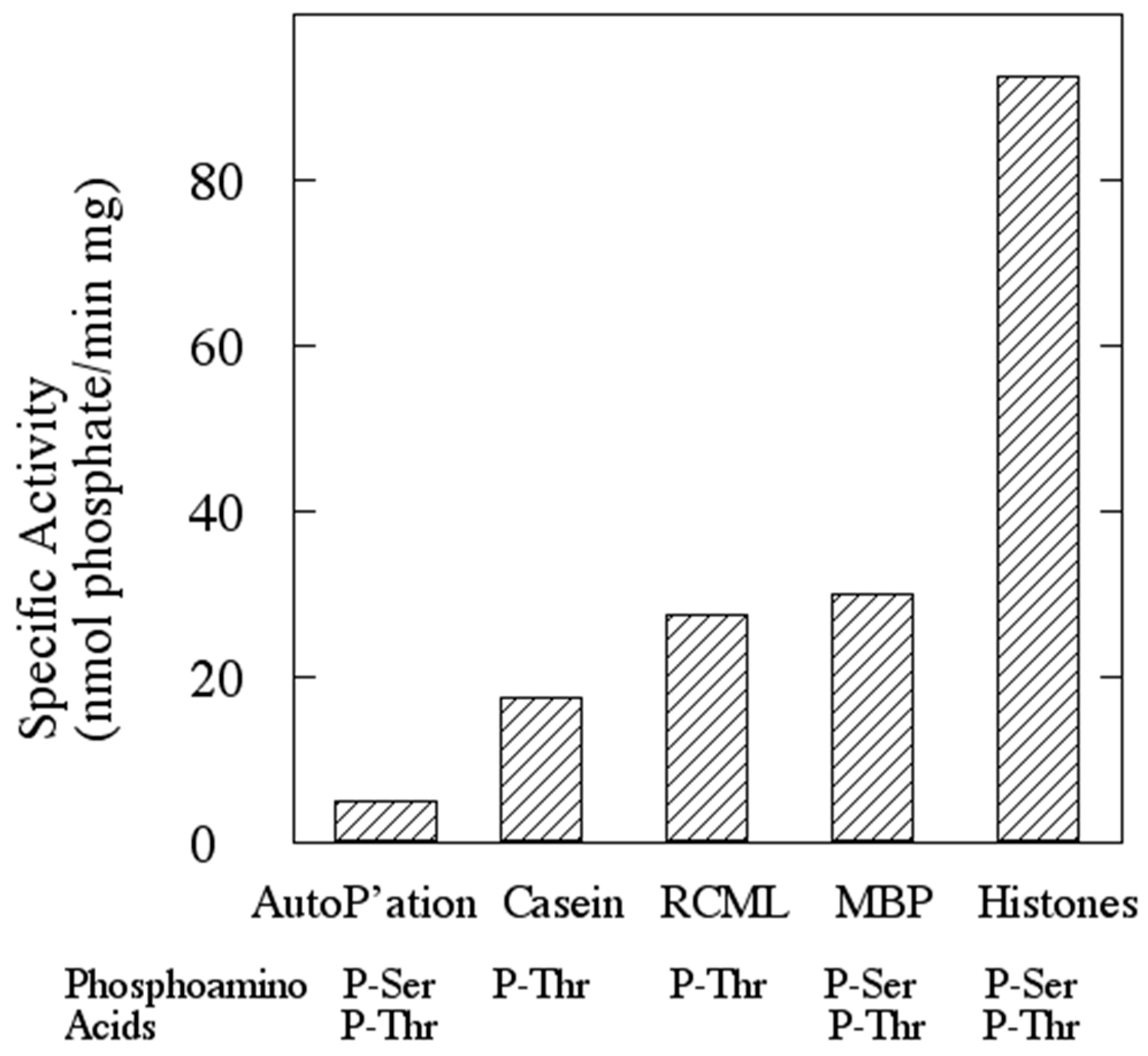
2.7. Phosphoamino Acid Analysis
2.8. Generation of Tryptic Peptides for MS Analysis
2.9. Clean-Up of Tryptic Peptides Using C18 Material
2.10. Analysis of Tryptic Peptides by Mass Spectrometry
3. Results and Discussion
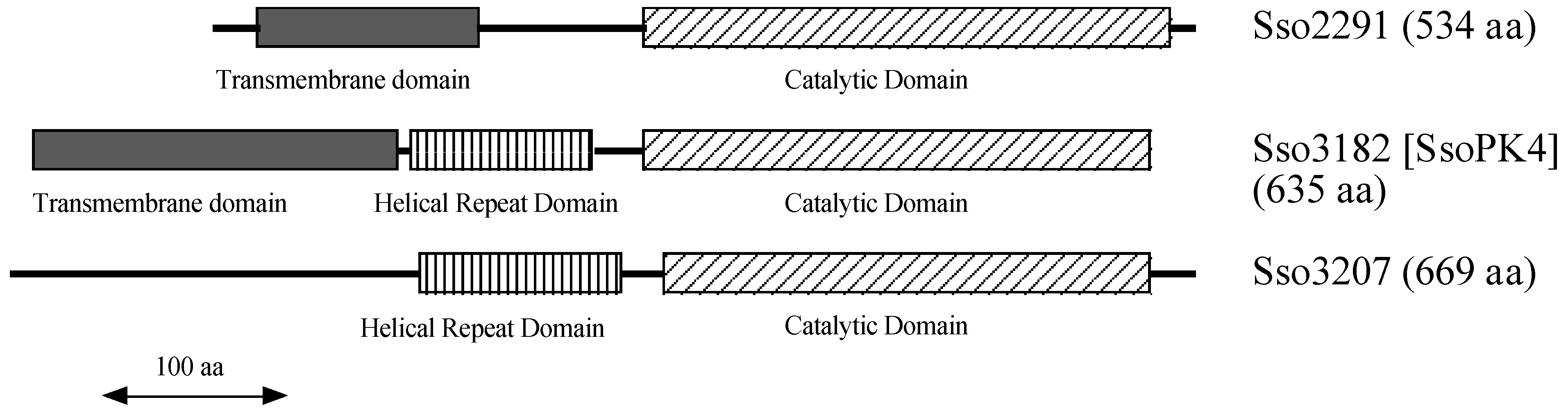
3.1. The Genome of Sulfolobus Solfataricus Encodes Three Deduced Typical ePKs

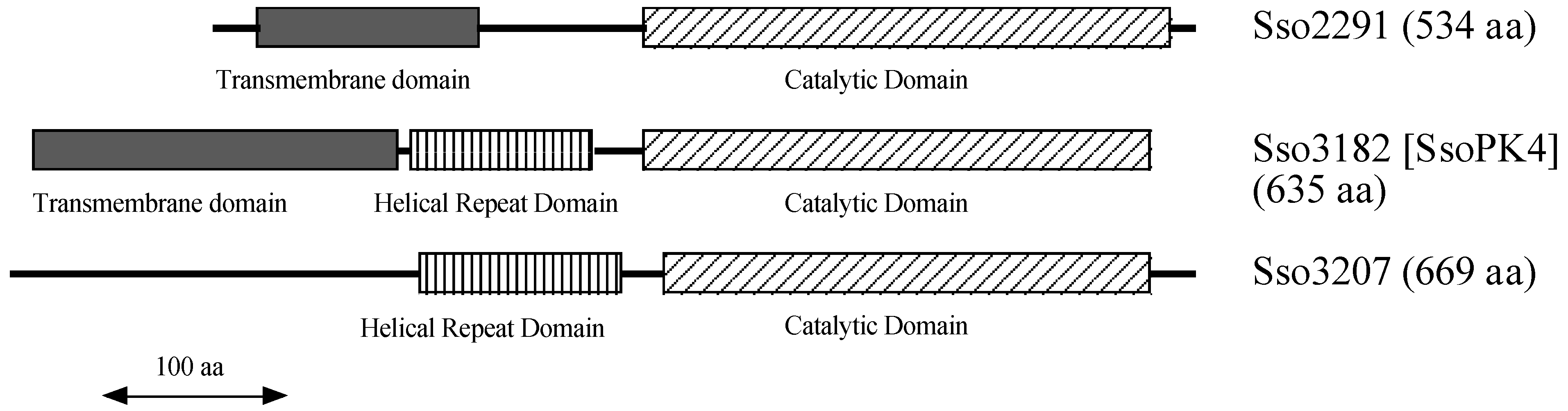
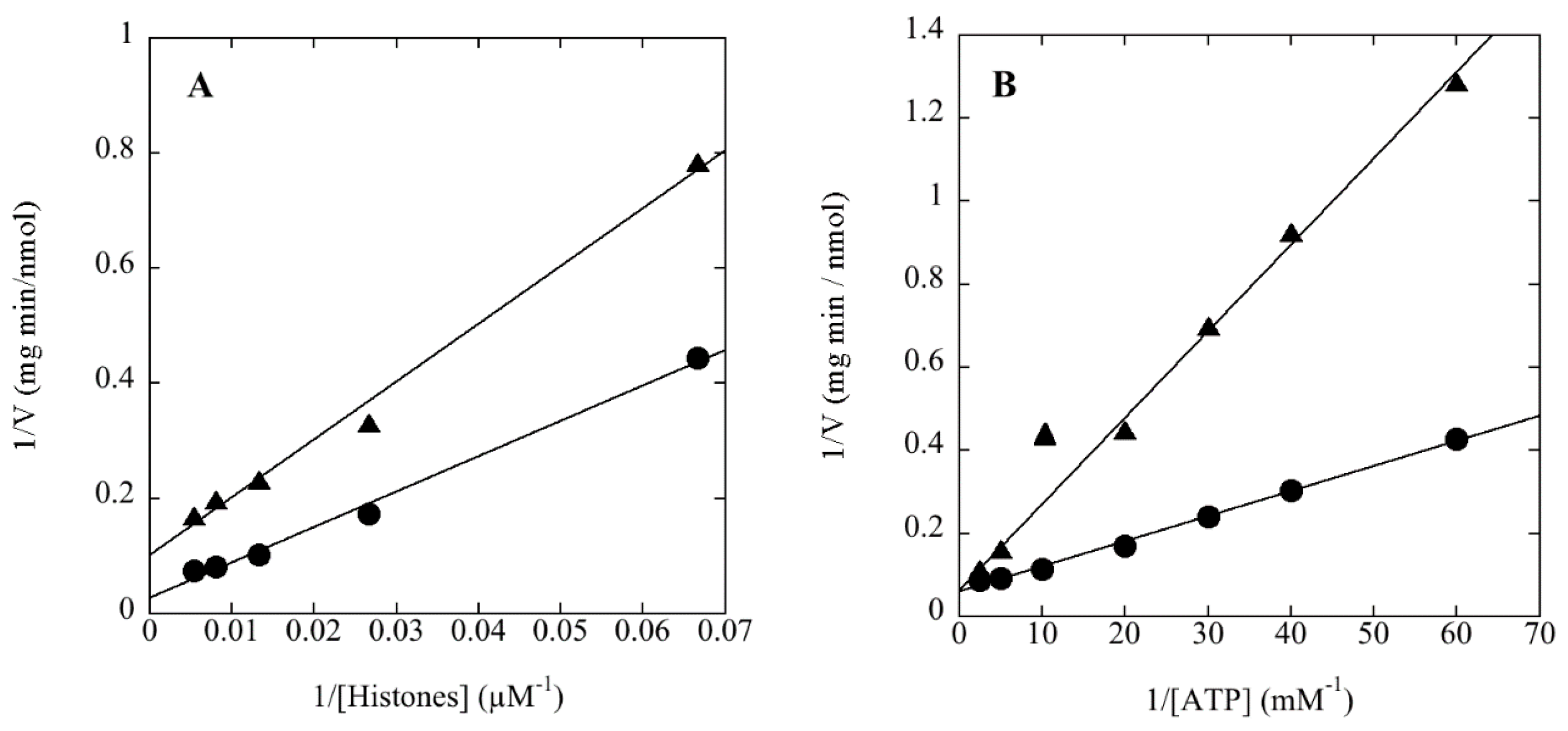
3.2. The Protein Product of ORF sso3182 Exhibits Protein-Serine/Threonine Kinase Activity
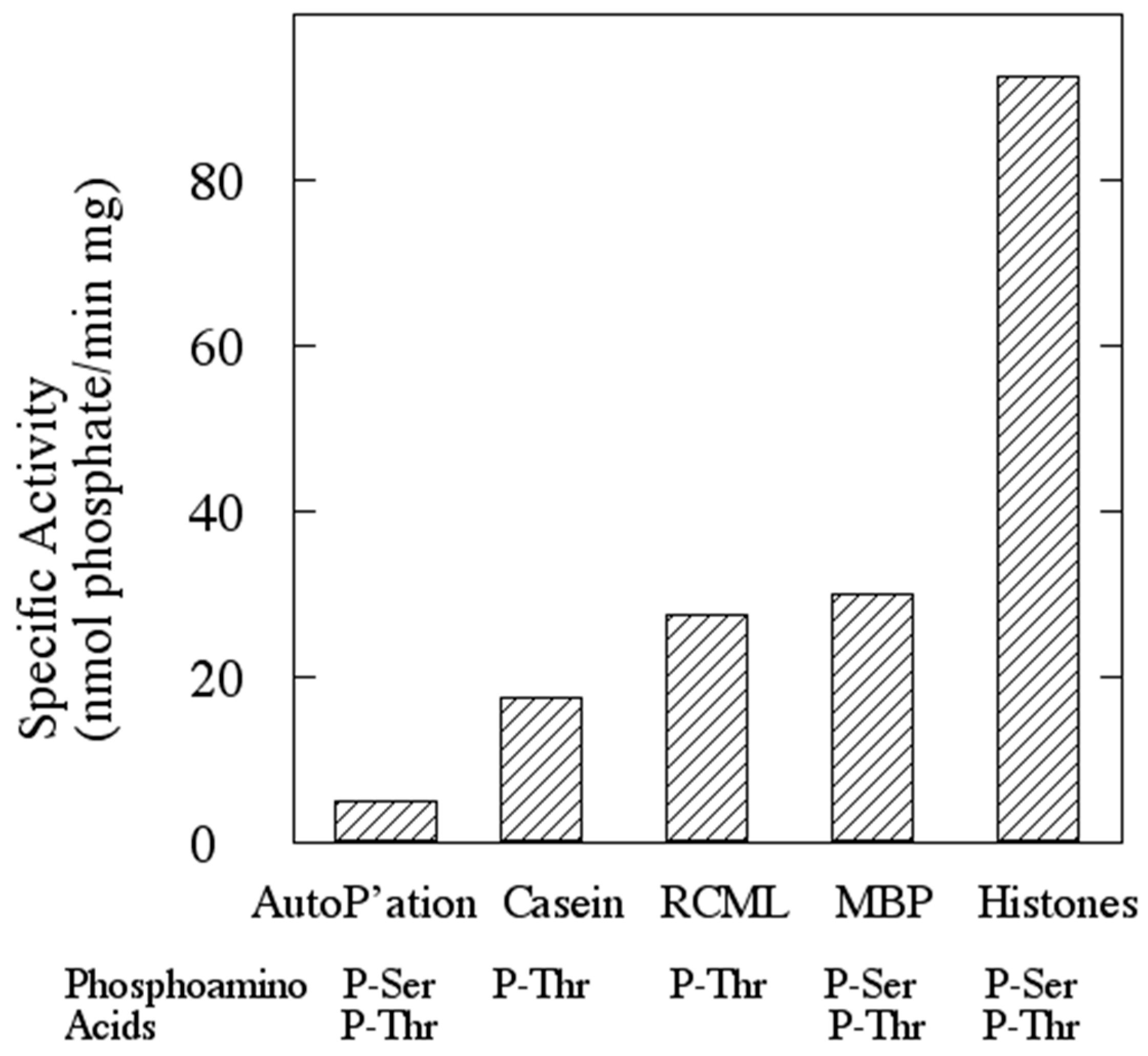
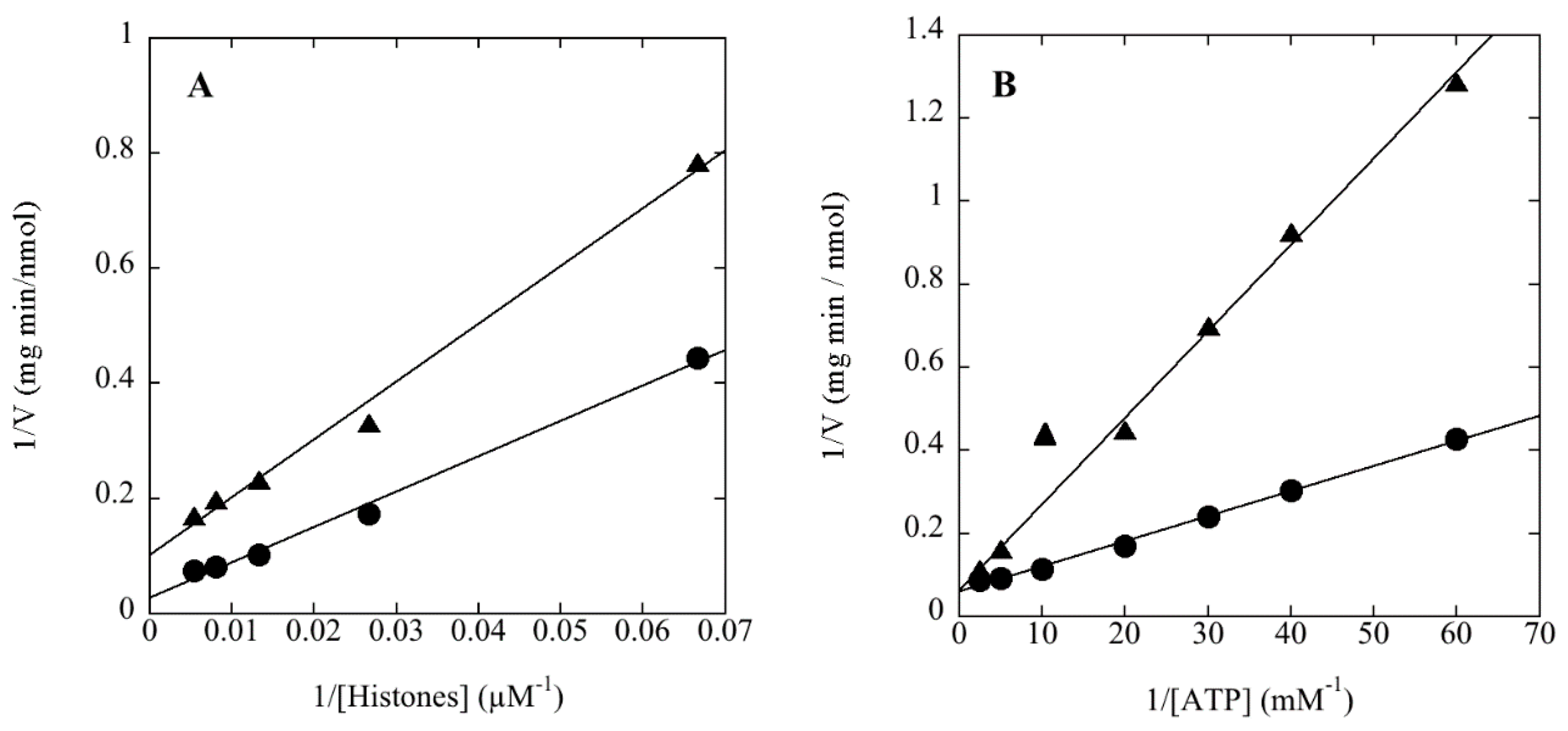
| Protein Kinase | Protein Substrate | CoAS-SCoA (1 mM) | Vmax (nmol/min mg) | Km (µM) |
|---|---|---|---|---|
| rSsoPK4(284–635) | Mixed histones | − | 16 | 220 |
| rSsoPK4(284–635) | MBP | − | 3.7 | 145 |
| rSsoPK4(284–635) | MBP | + | 4.8 | 48 |
| rSsoPK4(284–635) | aIF2α | − | 4.2 | 48 |
| rSsoPK4(284–635) | aIF2α | + | 2.9 | 6 |
| rSsoPK4(284–635) (T592D/T606D/S611D) | MBP | − | 4.2 | 17 |
| rSsoPK4(284–635) (T592D/T606D/S611D) | aIF2α | − | 1.9 | 4 |
3.3. rSsoPK4(284–635) Phosphorylates aIF2α in Vitro
| Phosphopeptide (z, m/z) | y-ion (m/z) | Δm (Da) | Predicted Residue(s) |
|---|---|---|---|
| #1 ( z = +2, m/z = 478.7) | 727.3 | 132.1 * | M |
| 640.5 | 86.8 | S | |
| 583.5 | 57.0 | G | |
| 470.3 | 113.2 | I/L | |
| 357.2 | 113.1 | I/L | |
| 274.3 | 82.9 | 2-Aminodehydrobutyrate | |
| n.a. | 274.3 | V + R | |
| #2 ( z = +2, m/z = 755.3) | 1114.4 | 298.2 * | I/L + Q/K + G |
| 984.5 | 129.9 | E or M ** | |
| 855.2 | 129.3 | E | |
| 741.5 | 113.7 | N | |
| 642.5 | 99.0 | V | |
| 527.5 | 115.0 | D | |
| 414.2 | 113.3 | I/L | |
| 345.2 | 69.0 | Dehydroalanine | |
| 246.1 | 99.1 | V | |
| n.a. | 246.1 | V + Q/K |

3.4. rSsoPK4(284–635) Catalyzes Its Own Phosphorylation
| Phosphopeptide (z, m/z) | b-ion (m/z) | y-ion (m/z) | y-Pi ion (m/z) | Δm (Da) | Predicted Residue(s) |
| #1 (z = +2, m/z = 656.4) | n.d. | 1148.5 | 1050.5 | 164.3 | Y |
| 277.1 | 1035.3 | 937.4 | 113.2 | I/L | |
| 392.3 | 920.4 | 822.4 | 115.1 | D | |
| 506.2 | 806.4 | 708.4 | 114.0 | N | |
| 593.3 | 719.4 | 621.4 | 87.0 | S | |
| 706.3 | 606.2 | 508.3 | 113.1 | I/L | |
| n.d. | 443.1 | 345.3 | 163.1 | Y | |
| n.d. | n.d. | n.d. | 443.2 | p(S + T + R) | |
| n.d. | n.d | n.d. | n.a. | n.a. | |
| n.d. | 175.0 | n.d. | 175.0 | R | |
| Phosphopeptide (z, m/z) | a-ion (m/z) | b-ion (m/z) | y-ion (m/z) | Δm (Da) | Predicted residue(s) |
| #2 (z = +2, m/z = 826.0) | 136.1 | 1291.7 | 136.1 | Y | |
| 235.1 | n.d. | 1192.6 | 99.1 | V | |
| n.d. | 378.2 | 1077.6 | 115.0 | D | |
| n.d. | 506.3 | 949.5 | 128.1 | Q/K | |
| n.d. | 620.3 | 835.5 | 114.0 | N | |
| n.d. | n.d. | 752.4 | 83.1 | 2-Amino-dehydrobutyrate | |
| n.d. | n.d. | 589.4 | 163.0 | Y | |
| n.d. | n.d. | 476.3 | 113.1 | I/L | |
| 1098.6 | 1126.6 | 329.2 | 147.1 | F | |
| 1211.6 | n.d. | 216.1 | 113.1 | I/L | |
| n.d. | n.d. | n.d. | 216.1 | Dehydroalanine + K/Q |

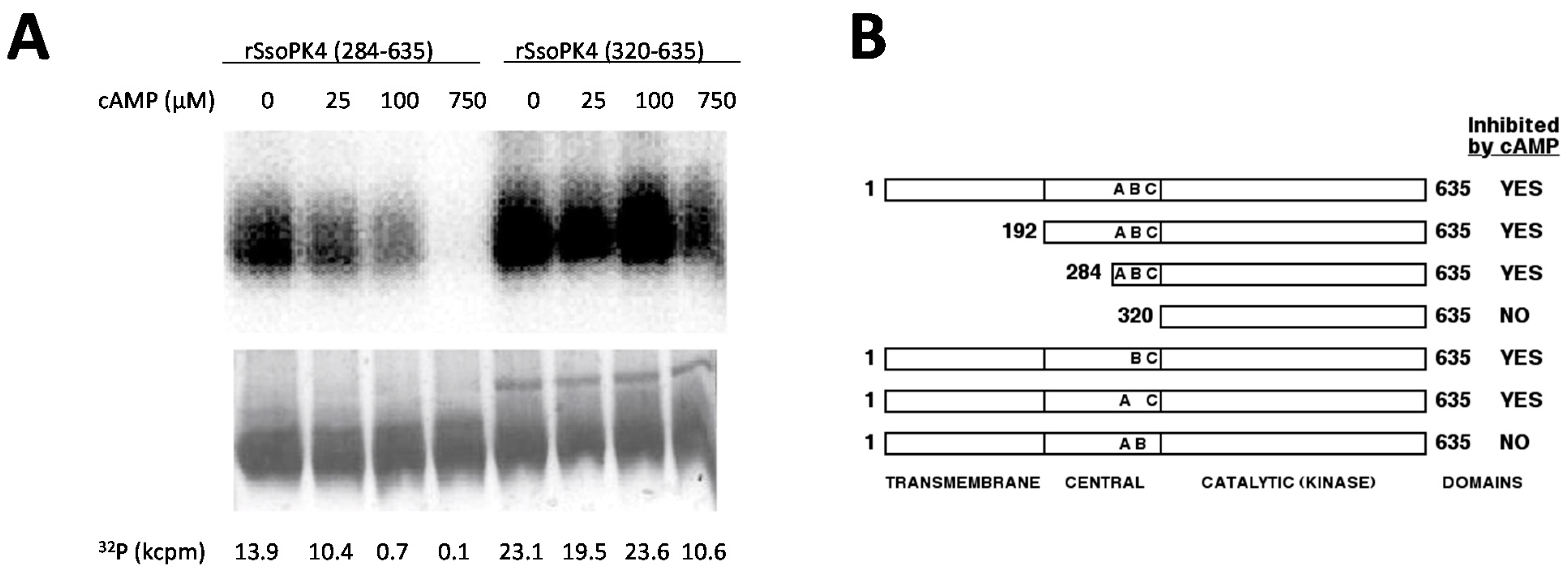
3.5. rSsoPK4 Is Inhibited by 3ʹ,5ʹ-cAMP in Vitro
| Nucleotide | Inhibitor | IC50 (µM) | Nucleotide | Inhibitor | IC50 (µM) |
|---|---|---|---|---|---|
| ADP | Y | 190 | NAD+ | N | n.d. |
| 5'AMP | Y | 920 | NADH | N | n.d. |
| 3ʹ-AMP | Y | 2100 | NADP+ | N | n.d. |
| 5ʹ-dAMP | N | n.d. | NADPH | N | n.d. |
| GTP | N | n.d. | FAD+ | N | n.d. |
| 5ʹ-GMP | N | n.d. | Thiamine-PPi | N | n.d. |
| 5ʹ-CMP | N | n.d. | CoASH | N | n.d. |
| 5ʹ-UMP | N | n.d. | Acetyl-CoA | N | n.d. |
| Phosphoadenosine phosphosulfate | N | n.d. | S-Adenosyl-Methionine | N | n.d. |
| Adenosine phosphosulfate | Y | 190 | S-Adenosyl-homocysteine | N | n.d. |
| 3ʹ,5ʹ-cAMP | Y | 50 | 2ʹ,3ʹ-cAMP | Y | 1550 |
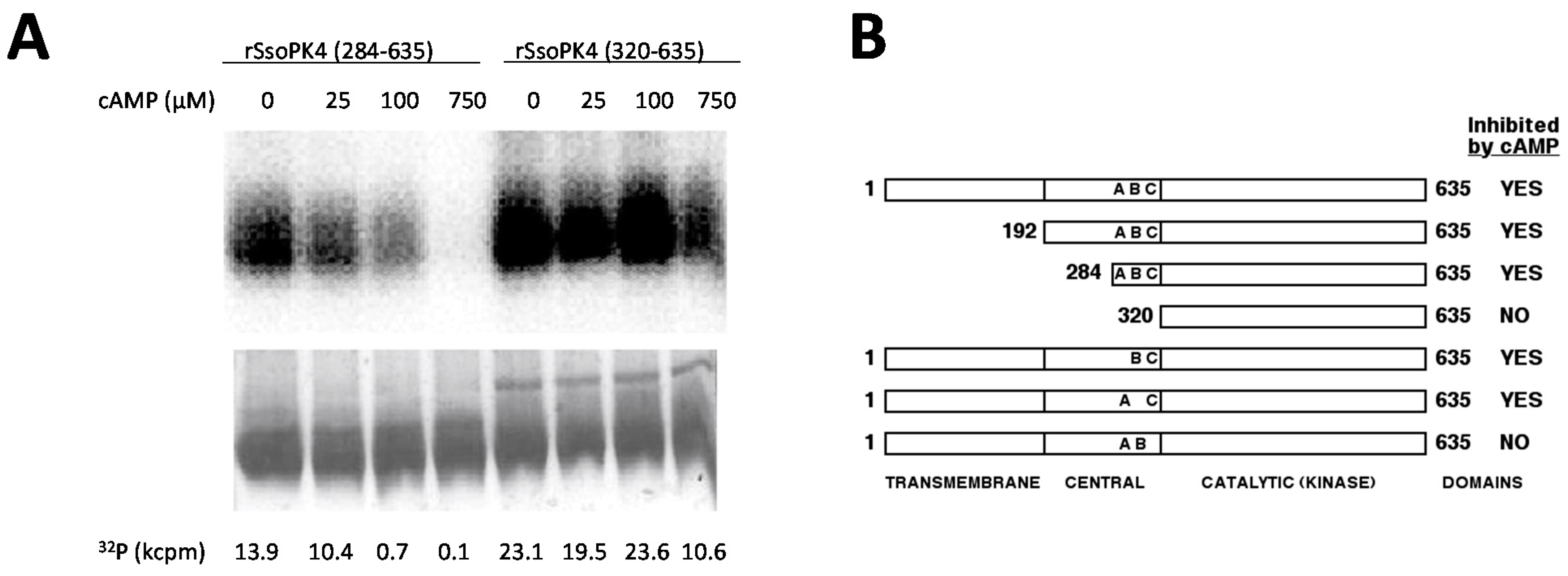
3.6. Residues 284-319 Are Necessary for Inhibition by 3',5'-cAMP
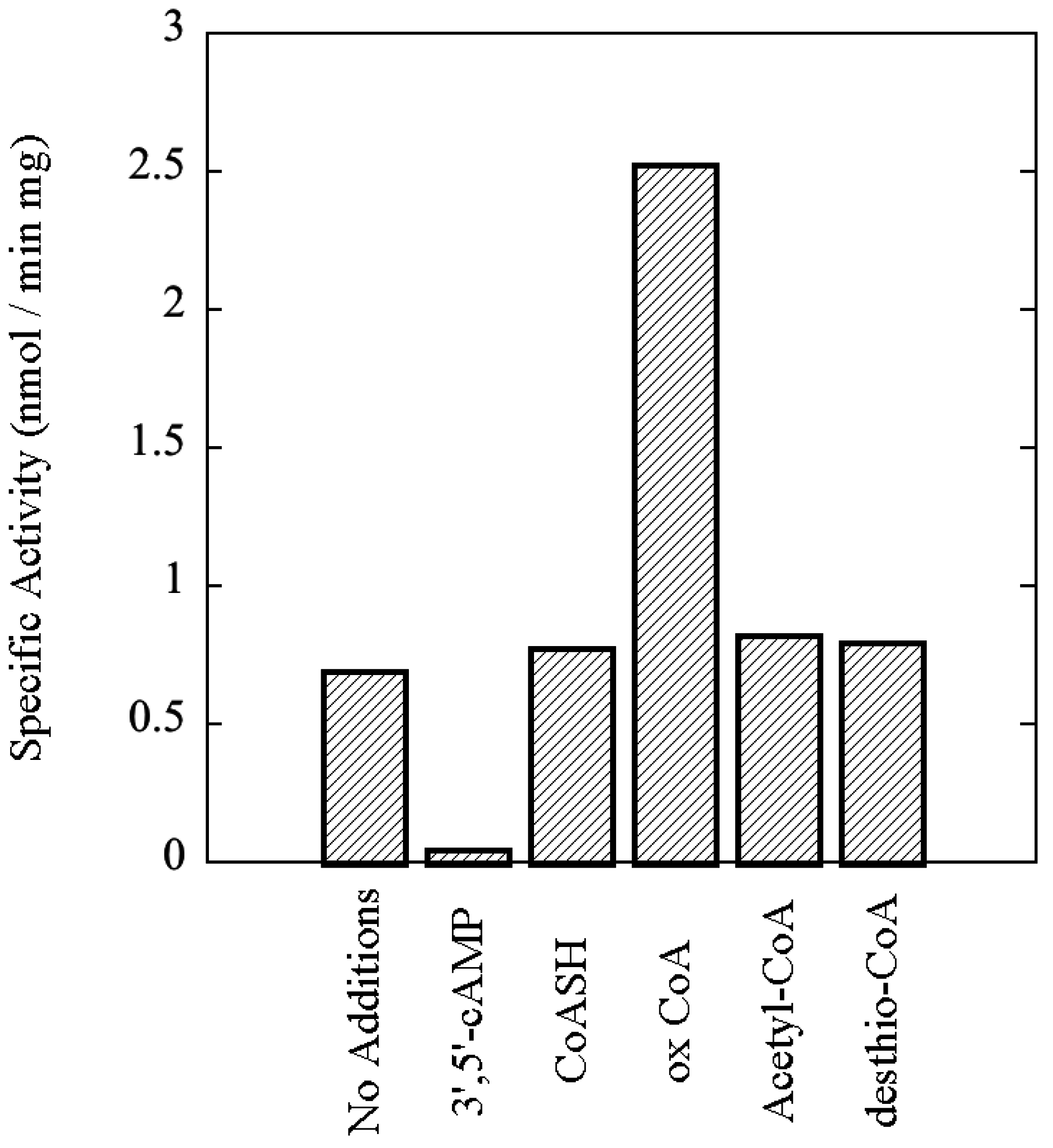
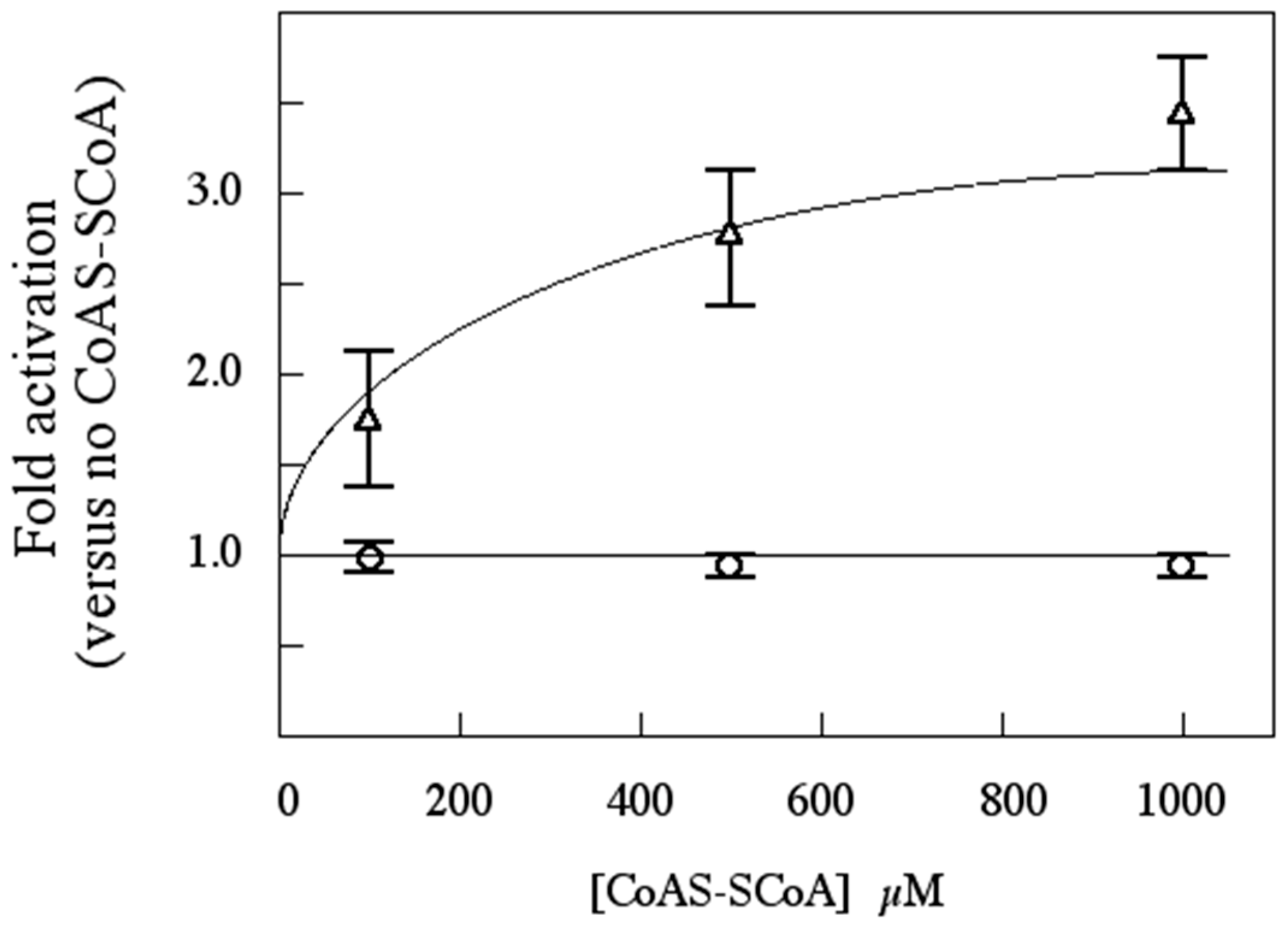
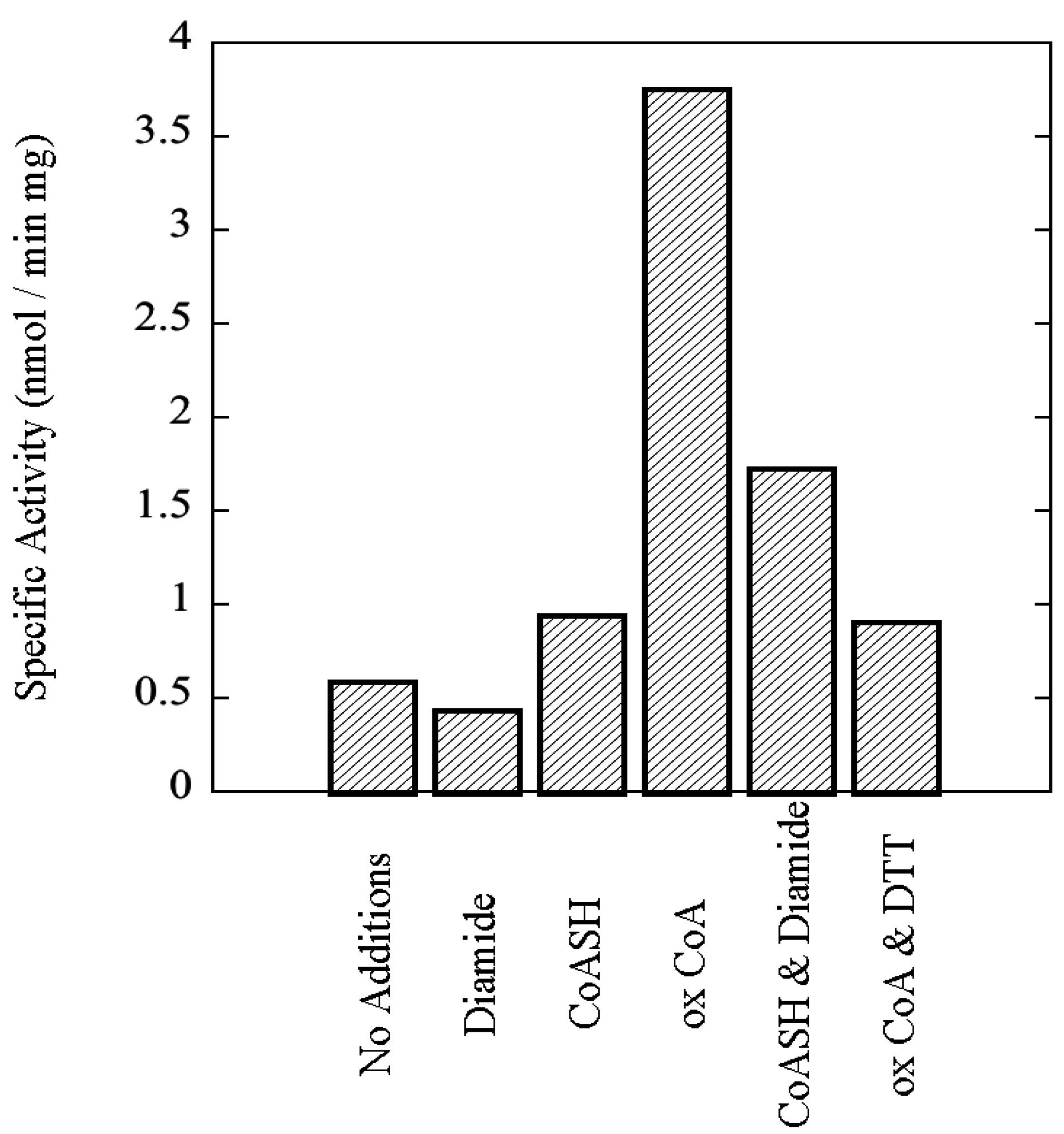
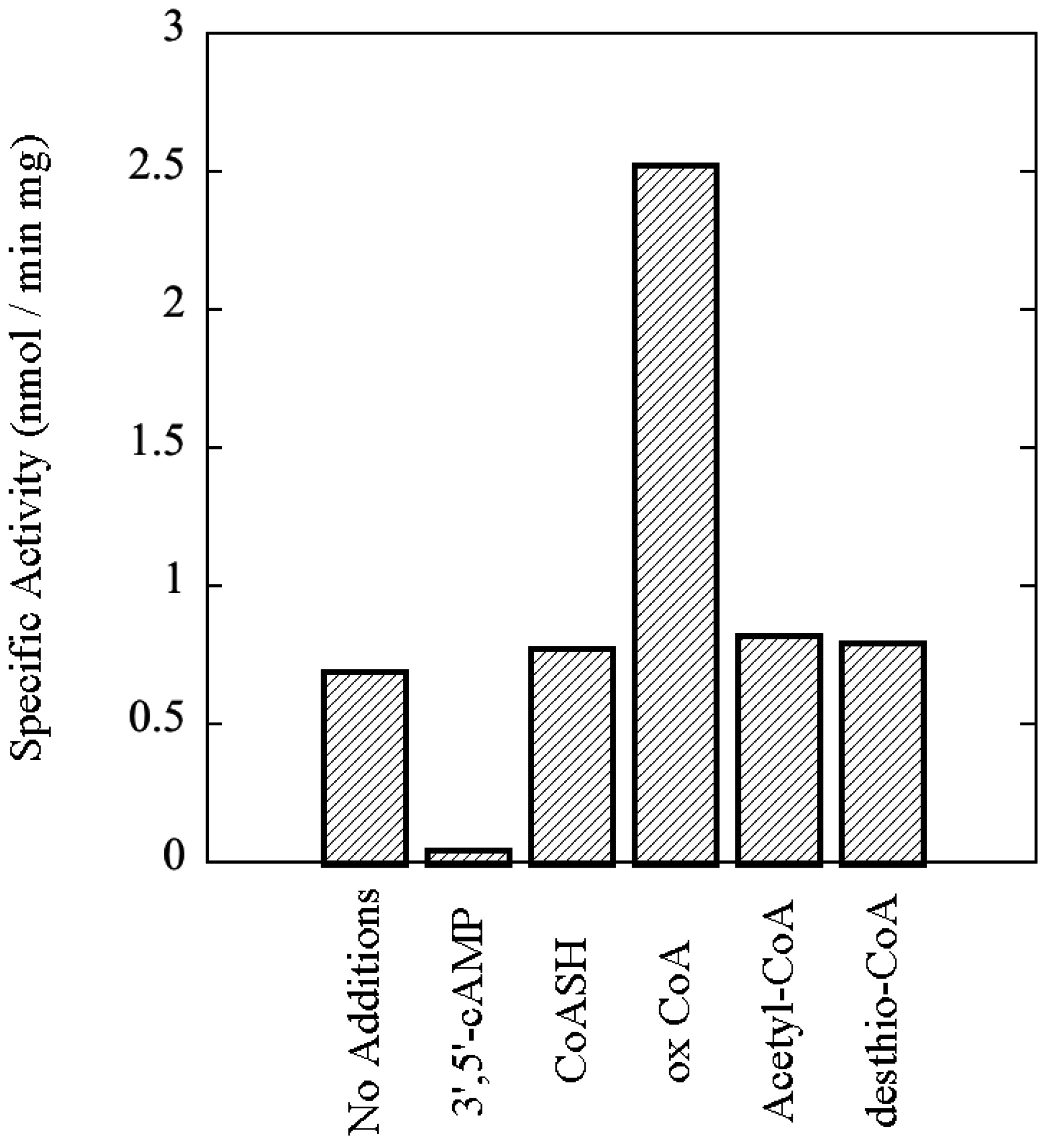
3.7. rSsoPK4(284–635) Is Activated by Oxidized CoA
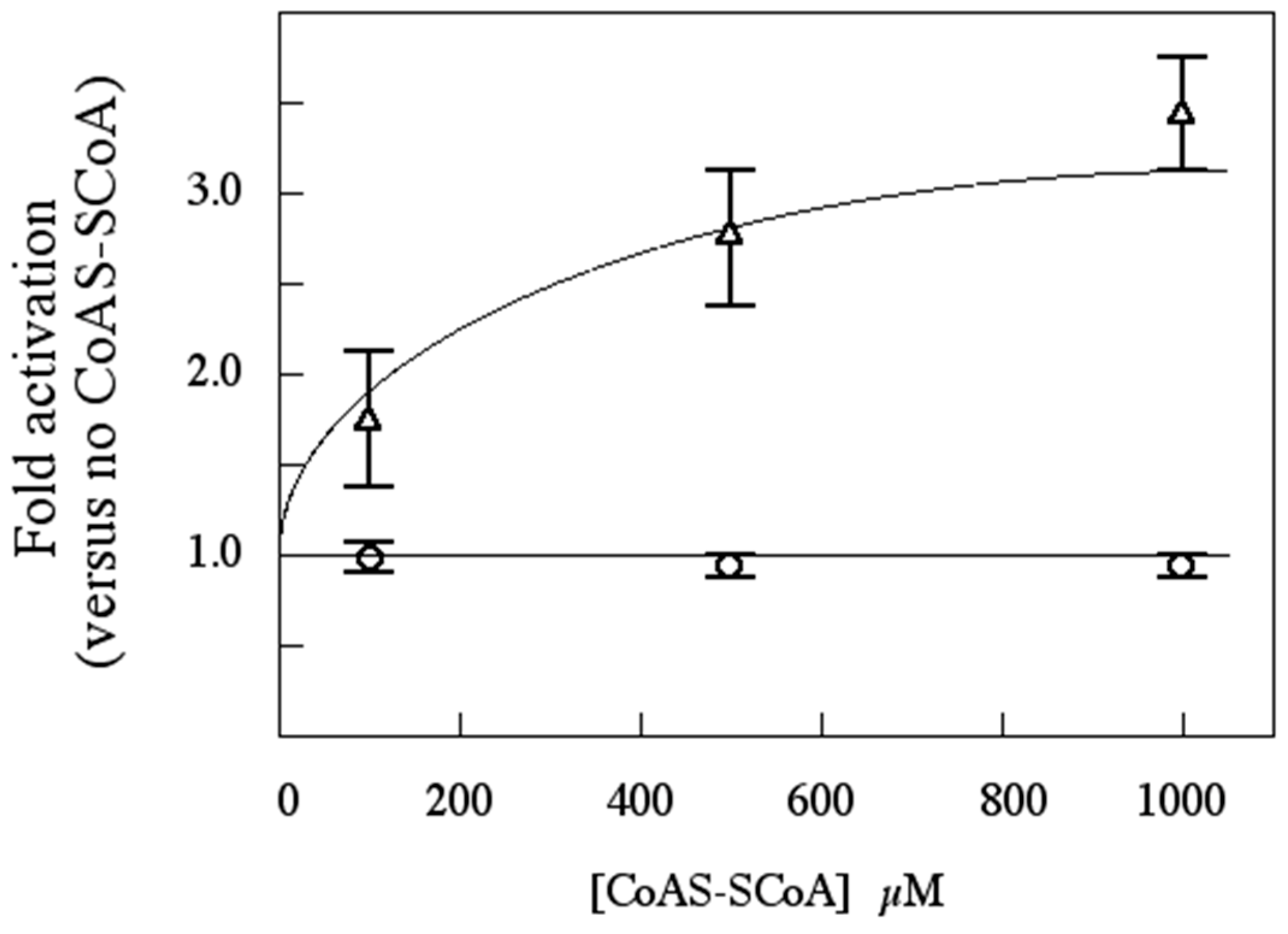
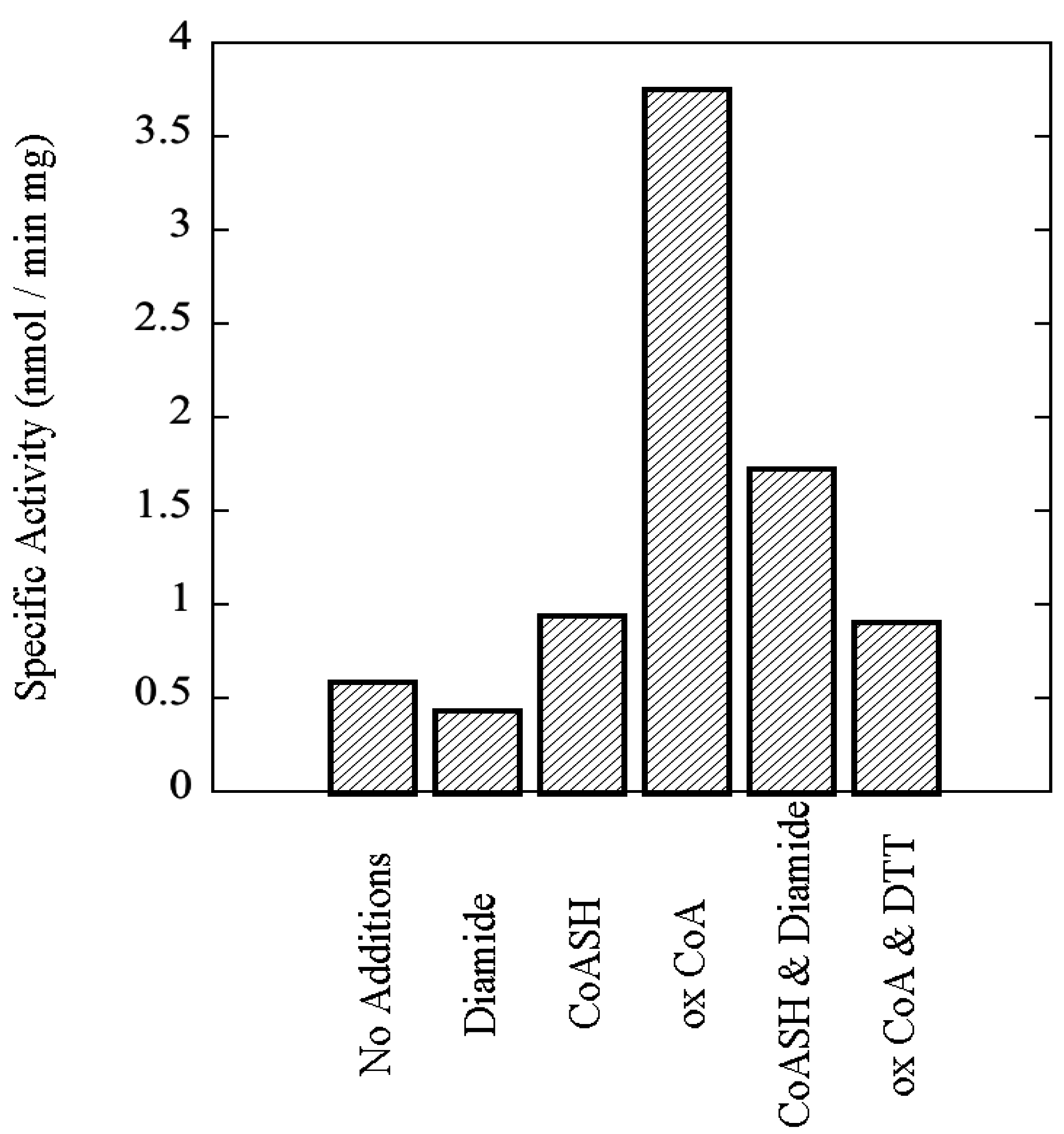
3.8. Functional Impact of Autophosphorylation
3.9. Is SsoPK4 an aIF2α Protein Kinase?
| Archaeon | Sequence | Position of Residue in Bold |
|---|---|---|
| Sulfolobus solfataricus | glitvrtnep | 184 |
| Sulfolobus acidocaldarius | dvislrtidp | 190 |
| Sulfolobus tokadaii | eivtlrssdp | 190 |
| Pyrobaculum aerophilum | kivsvegdgv | 191 |
| Pyrobaculum islandicum | kavsvegdga | 191 |
| Aeropyrum pernix | tlrsmagdgv | 203 |
| Thermoproteus neutrophilus | kavsvegdga | 191 |
| Igniccocus hospitalis | ilqsfapdgv | 191 |
3.10. Does 3',5ʹ-cAMP Regulate SsoPK4 in Vivo?
3.11. Is CoAS-SCoA a Plausible Physiological Activator for SsoPK4?
4. Conclusions
How Did S. solfataricus and Other Archaeons Acquire Typical ePKs such as SsoPK4?
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bray, D. Proteins as computational elements in living cells. Nature 1995, 376, 307–312. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef]
- Wurgler-Murphy, S.M.; King, D.M.; Kennelly, P.J. The phosphorylation site database: A guide to the serine-, threonine-, and/or tyrosine- phosphorylated proteins in prokaryotic organisms. Proteomics 2004, 4, 1562–1570. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S.D. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.J.; Aravind, L.; Koonin, E.V. Novel families of putative protein kinases in Bacteria and Archaea: Evolution of the ‘eukaryotic’ protein kinase superfamily. Genome Res. 1998, 8, 1038–1047. [Google Scholar] [PubMed]
- Shi, L.; Potts, M.P.; Kennelly, P.J. The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: A family portrait. FEMS Microbiol. Rev. 1998, 22, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Aravind, L.; Schultz, J.; Bork, P.; Koonin, E.V. Eukaryotic signaling domain homologues in Archaea and Bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 1999, 289, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Taylor, S.S.; Zhai, Y.; Ventner, J.C.; Manning, G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007, 5, 467–478. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- LaRonde, N.A. The ancient microbial RIO kinases. J. Biol. Chem. 2014, 289, 9488–9492. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2 kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and simple method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, G.; Steck, T.L.; Wallace, D.F.H. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry 1971, 10, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Ray, W.K.; Keith, S.M.; DeSantis, A.M.; Hunt, J.P.; Larson, T.J.; Helm, R.F.; Kennelly, P.J. A phosphohexomutase from the archaeon Sulfolobus solfataricus is covalently modified by phosphorylation on serine. J. Bacteriol. 2005, 187, 4270–4275. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Reimann, E.M. Assay of cAMP-dependent protein kinases. Meth. Enzymol. 1971, 38, 287–290. [Google Scholar]
- Lower, B.H.; Kennelly, P.J. Open reading frame sso2387 from the archaeon Sulfolobus solfataricus encodes a polypeptide with protein-serine kinase activity. J. Bacteriol. 2003, 185, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Baxter-Gabbard, K.L. A simple method for the large-scale preparation of sucrose gradients. FEBS Lett. 1972, 20, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Kamps, M.P.; Sefton, B.M. Acid and base hydrolysis of phosphoproteins bound to immobilon facilitates analysis of phosphoamino acids in gel-fractionated proteins. Anal. Biochem. 1989, 176, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Pappin, D.J.C.; Hojrup, P.; Bleasby, A.J. Rapid identification of proteins by mass peptide fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Esser, D.; Pham, T.K.; Reimann, J.; Albers, S.V.; Siebers, B.; Wright, P.C. Change of carbon source causes dramatic effects in the phospho-proteome of the archaeon Sulfolobus solfataricus. J. Proteome Res. 2012, 11, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- She, Q.; Singh, R.K.; Confalonieri, F.; Zivanovic, Y.; Allard, G.; Awayez, M.J.; Chan-Weiher, C.C.; Clausen, I.G.; Curtis, B.A.; de Moors, A.; et al. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc. Natl. Acad. Sci. USA 2001, 98, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Kennelly, P.J. Protein ser/thr/tyr phosphorylation in the Archaea. J. Biol. Chem. 2014, 289, 9480–9487. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.W.B.; Cosentino, G.P.; Donze, O.; Sonenburg, N. The kinase insert domain of interferon-induced protein kinase PKR is required for activity but not for interaction with pseudosubstrate K3L. J. Biol. Chem. 1996, 271, 24526–24533. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, J.J.; Santoyo, J.; de Haro, C. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2α kinase. Eur. J. Biochem. 1999, 265, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Groves, M.R.; Barford, D. Topological characteristics of helical repeat proteins. Curr. Opin. Struct. Biol. 1999, 9, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Knighton, D.R.; Zheng, J.; Sowadski, J.M.; Gibbs, C.S.; Zoller, M.J. A template for the protein kinase family. Trends Biochem. Sci. 1993, 18, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Lindberg, R.A. Use of degenerate oligonucleotide probes to identify clones that encode protein kinases. Meth. Enzymol. 1991, 200, 525–532. [Google Scholar] [PubMed]
- Lower, B.H.; Potters, M.B.; Kennelly, P.J. A phosphoprotein from the archaeon Sulfolobus solfataricus with protein-serine/threonine kinase activity. J. Bacteriol. 2004, 186, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Cameron, A.J.; Buckel, S.; Kennelly, P.J. Isolation and cloning of a protein-serine/threonine phosphatase from an archaeon. J. Bacteriol. 1995, 177, 6510–6517. [Google Scholar] [PubMed]
- Allen, G.S.; Frank, J. Structural insights on the translation initiation complex: Ghosts of a universal initiation complex. Mol. Microbiol. 2007, 63, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Maone, E.; di Stefano, M.; Berardi, A.; Benelli, D.; Marzi, S.; La Teana, A.; Londei, P. Functional analysis of the translation factor aIF2/5B in the thermophilic archaeon Sulfolobus solfataricus. Mol. Microbiol. 2007, 65, 700–713. [Google Scholar] [CrossRef] [PubMed]
- La Teana, A.; Benelli, D.; Londei, P.; Blasi, U. Translation initiation in the crenarchaeon Sulfolobus solfataricus: Eukaryotic features but bacterial route. Biochem. Soc. Trans. 2013, 41, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Oshawa, A.; Saito, S.; Kimura, M. In vitro phosphorylation of initiation factor 2α (aIF2α) from hyperthermophilic archaeon Pyrococcus horikoshii OT3. J. Biochem. 2004, 135, 479–485. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.W.; Weitzman, P.D.; Morris, J.G. Oxidation of a variety of natural electron donors by the thiol-oxidising agent diamide. FEBS Lett. 1970, 10, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Staschke, K.A.; Zhu, S.; Wek, R.C. Inhibitory sequences in the N-terminus of the double-stranded-RNA-dependent protein kinase, PKR, are important for regulating phosphorylation of eukaryotic initiation factor 2α (eIF2α). Eur. J. Biochem. 2001, 268, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Kudlicki, W.; Fullilove, S.; Read, R.; Kramer, G.; Hardesty, B. Identification of spectrin-related peptides associated with the reticulocyte heme-controlled α subunit of eukaryotic initiation factor 2 kinase and of a Mr 95,000 peptide that appears to be the catalytic domain. J. Biol. Chem. 1987, 262, 9695–9701. [Google Scholar] [PubMed]
- Proud, C.G.; Colthurst, D.R.; Ferrari, S.; Pinna, L.A. The substrate specificity of protein kinases which phosphorylate the α subunit of eukaryotic initiation factor 2. Eur. J. Biochem. 1991, 195, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, E.A.; O’Brien, L.; Henshaw, E.C.; Panniers, R. Purified and characterization of eukaryotic initiation factor (eIF)-2α kinases from Ehrlich ascites tumor cells. J. Biol. Chem. 1993, 268, 12552–12559. [Google Scholar] [PubMed]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Fagard, R.; London, I.M. Relationship between phosphorylation and activity of heme-regulated eukaryotic initiation factor 2α kinase. Proc. Natl. Acad. Sci. USA 1981, 78, 868–870. [Google Scholar] [CrossRef]
- Romano, P.R.; Garcia-Barrio, M.T.; Zhang, X.; Wang, Q.; Taylor, D.R.; Zhang, F.; Herring, C.; Mathews, M.B.; Qin, J.; Hinnebusch, A.G. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell. Biol. 1998, 18, 2282–2297. [Google Scholar] [PubMed]
- Thomis, D.C.; Samuel, C.E. Mechanism of interferon action: Characterization of intermolecular autophosphorylation of PKR, the interferon-inducible, RNA-dependent protein kinase. J. Virol. 1995, 69, 5195–5198. [Google Scholar] [PubMed]
- Bauer, B.N.; Rafie-Kolpin, M.; Lu, L.; Han, A.; Chen, J.-J. Multiple autophosphorylation is essential for the formation of the active and stable homodimer of heme-regulated eIF2α kinase. Biochemistry 2001, 40, 11543–11551. [Google Scholar] [CrossRef] [PubMed]
- Padyana, A.K.; Qiu, H.; Roll-Mecak, A.; Hinnebusch, A.G.; Burley, S.K. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2α protein kinase GCN2. J. Biol. Chem. 2005, 280, 29289–29299. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Soufi, B.; Gnad, F.; Jensen, P.R.; Petranovic, D.; Mann, M.; Mijakovic, I.; Macek, B. The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL 1403 reveals multiply phosphorylated proteins. Proteomics 2008, 8, 3486–3493. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Tuch, B.B.; Villen, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; de Godoy, L.M.F.; Cox, J.; Neuhauser, N.; Ren, S.; Olsen, J.V.; Mann, M. High-accuracy identification and bioinformatics analysis of in vivo protein phosphorylation sites in yeast. Proteomics 2009, 9, 4642–4652. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.R.; Levy, E.D.; Michnick, S.W. Weak functional constraints on phosphoproteomes. Trends Genetics 2009, 25, 193–197. [Google Scholar] [CrossRef]
- Freschi, L.; Ossei, M.; Landry, C.R. Functional divergence and evolutionary turnover in mammalian phosphoproteomes. PLoS Genetics 2014, 10, e1004062. [Google Scholar] [CrossRef] [PubMed]
- Zhulin, I.B.; Nikolskaya, A.N.; Galperin, M.Y. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in Bacteria and Archaea. J. Bacteriol. 2003, 185, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yakunin, A.F.; Kuznetsova, E.; Busso, D.; Pufan, R.; Proudfoot, M.; Kim, R.; Kim, S.-H. Structural and functional characterization of a novel phosphodiesterase from Methanococcus jannaschii. J. Biol. Chem. 2004, 279, 31854–31862. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Lange, C.; Soppa, J. Transcriptome and cAMP oscillations in an archaeal cell cycle. BMC Cell Biol. 2007, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Leichtling, B.H.; Rickenberg, H.W.; Seely, R.J.; Fahrney, D.E.; Pace, N.R. The occurrence of cyclic AMP in archaebacteria. Biochem. Biophys. Res. Commun. 1986, 136, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Hummel, C.S.; Lancaster, K.M.; Crane, E.J., III. Determination of coenzyme A levels in Pyrococcus furiosus and other Archaea: Implications for a general role for coenzyme A in thermophiles. FEMS Microbiol. Lett. 2005, 252, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Ogino, M.; Orita, I.; Nakamura, S.; Imanaka, T.; Fukui, T. Characterization of NADH oxidase/NADPH polysulfide oxidoreductase and its unexpected participation in oxygen sensitivity in an anaerobic hyperthermophilic archaeon. J. Bacteriol. 2010, 192, 5192–5202. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.-G.; Kim, S.-H. Global extent of horizontal gene transfer. Proc. Natl. Acad. Sci. USA 2007, 104, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Kort, J.C.; Esser, D.; Pham, T.K.; Noirel, J.; Wright, P.C.; Seibers, B. A cool tool for hot and sour Archaea: Proteomics in Sulfolobus solfataricus. Proteomics 2013, 13, 2831–2850. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, W.K.; Potters, M.B.; Haile, J.D.; Kennelly, P.J. Activation of SsoPK4, an Archaeal eIF2α Kinase Homolog, by Oxidized CoA. Proteomes 2015, 3, 89-116. https://doi.org/10.3390/proteomes3020089
Ray WK, Potters MB, Haile JD, Kennelly PJ. Activation of SsoPK4, an Archaeal eIF2α Kinase Homolog, by Oxidized CoA. Proteomes. 2015; 3(2):89-116. https://doi.org/10.3390/proteomes3020089
Chicago/Turabian StyleRay, William K., Mark B. Potters, January D. Haile, and Peter J. Kennelly. 2015. "Activation of SsoPK4, an Archaeal eIF2α Kinase Homolog, by Oxidized CoA" Proteomes 3, no. 2: 89-116. https://doi.org/10.3390/proteomes3020089
APA StyleRay, W. K., Potters, M. B., Haile, J. D., & Kennelly, P. J. (2015). Activation of SsoPK4, an Archaeal eIF2α Kinase Homolog, by Oxidized CoA. Proteomes, 3(2), 89-116. https://doi.org/10.3390/proteomes3020089