
The Cutting Edge of Affinity Electrophoresis Technology



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
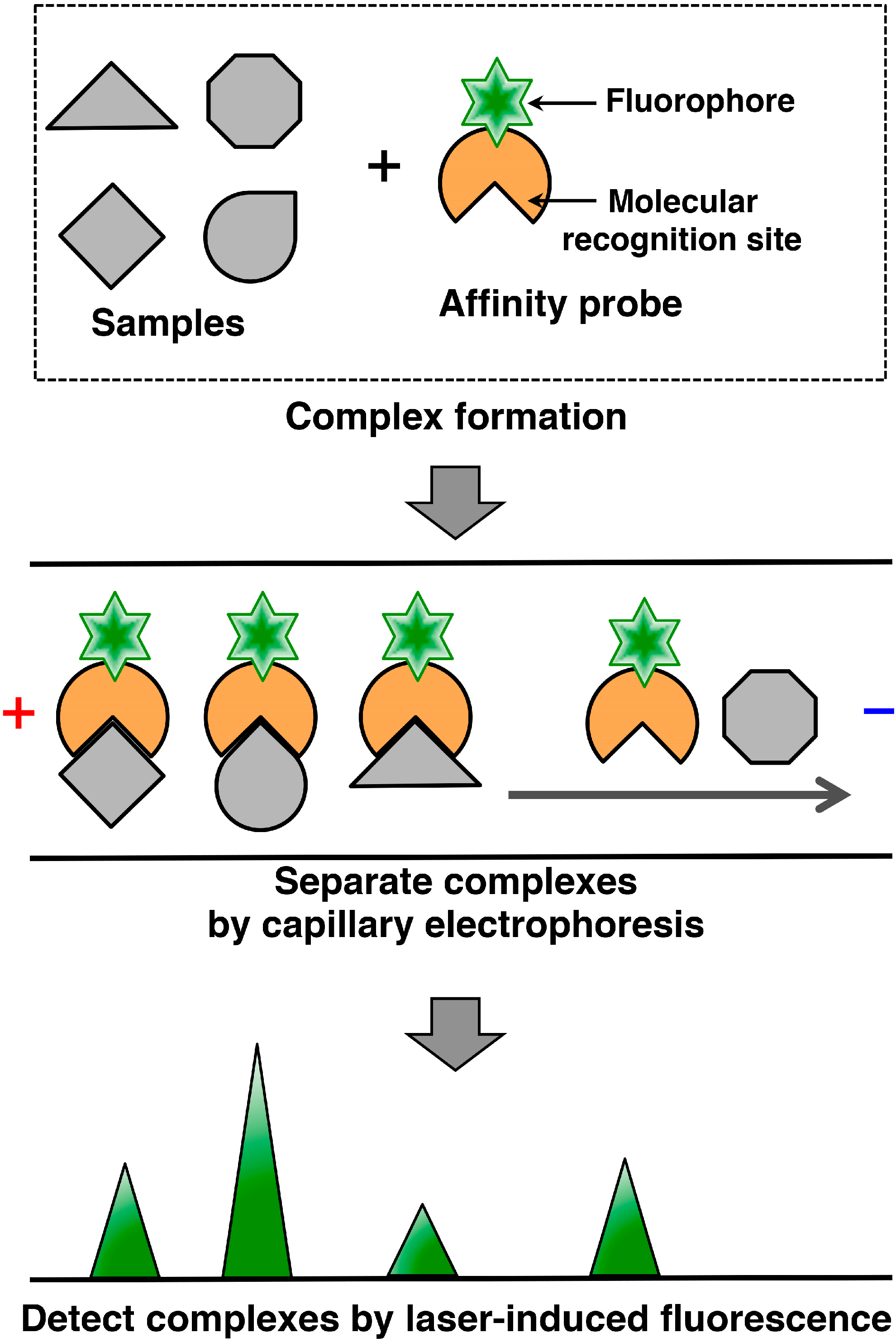
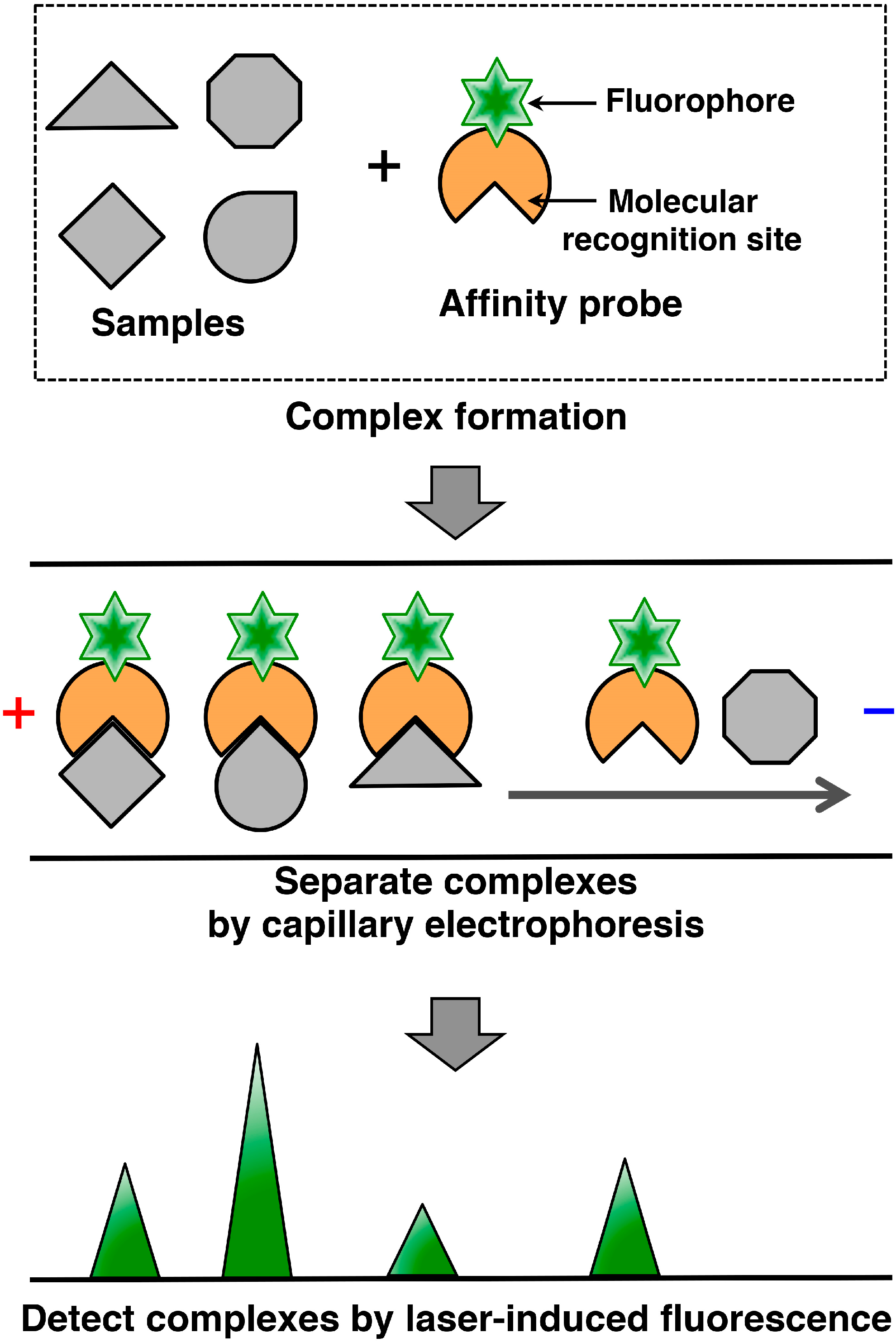
2. Capillary Affinity Electrophoresis
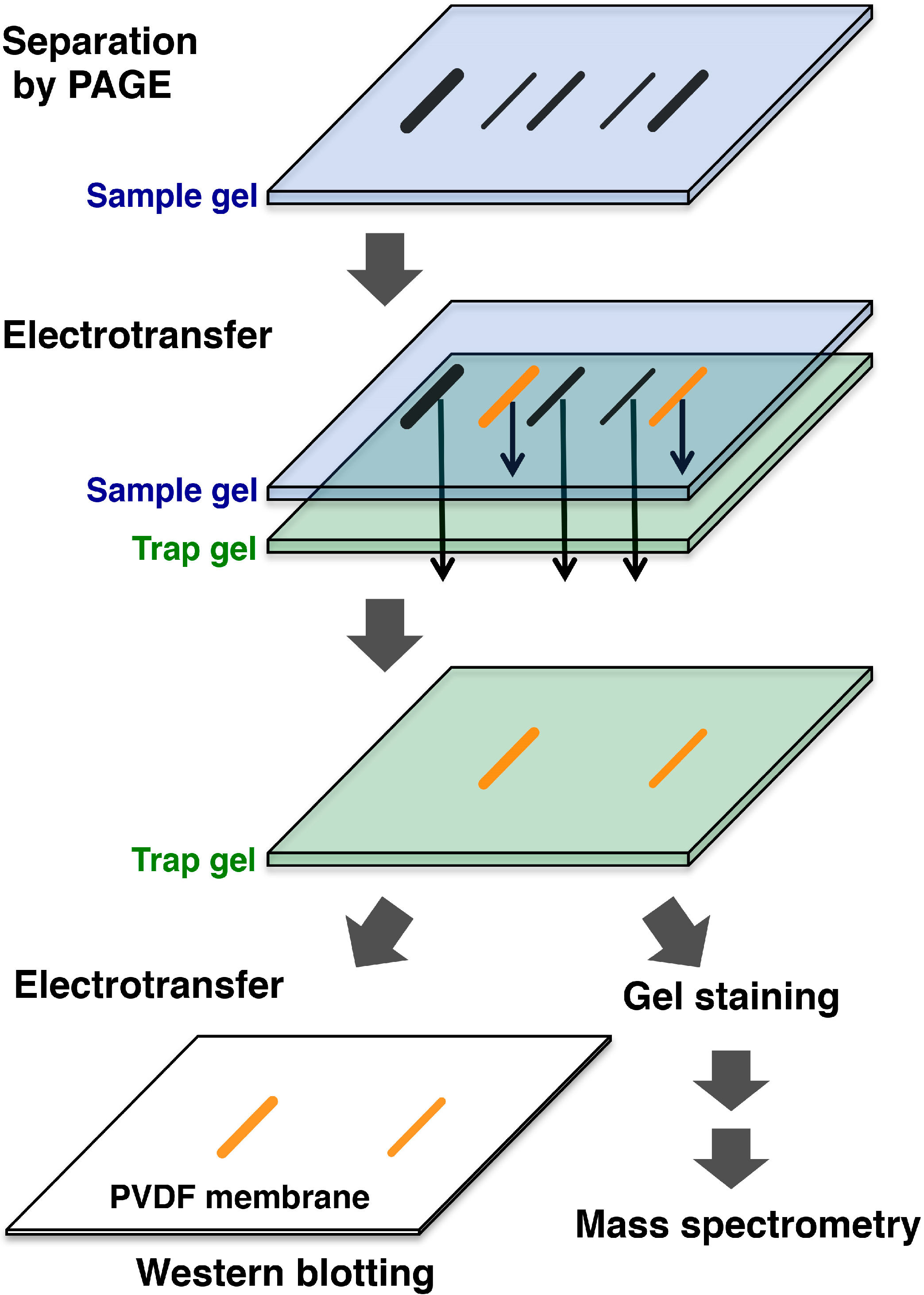
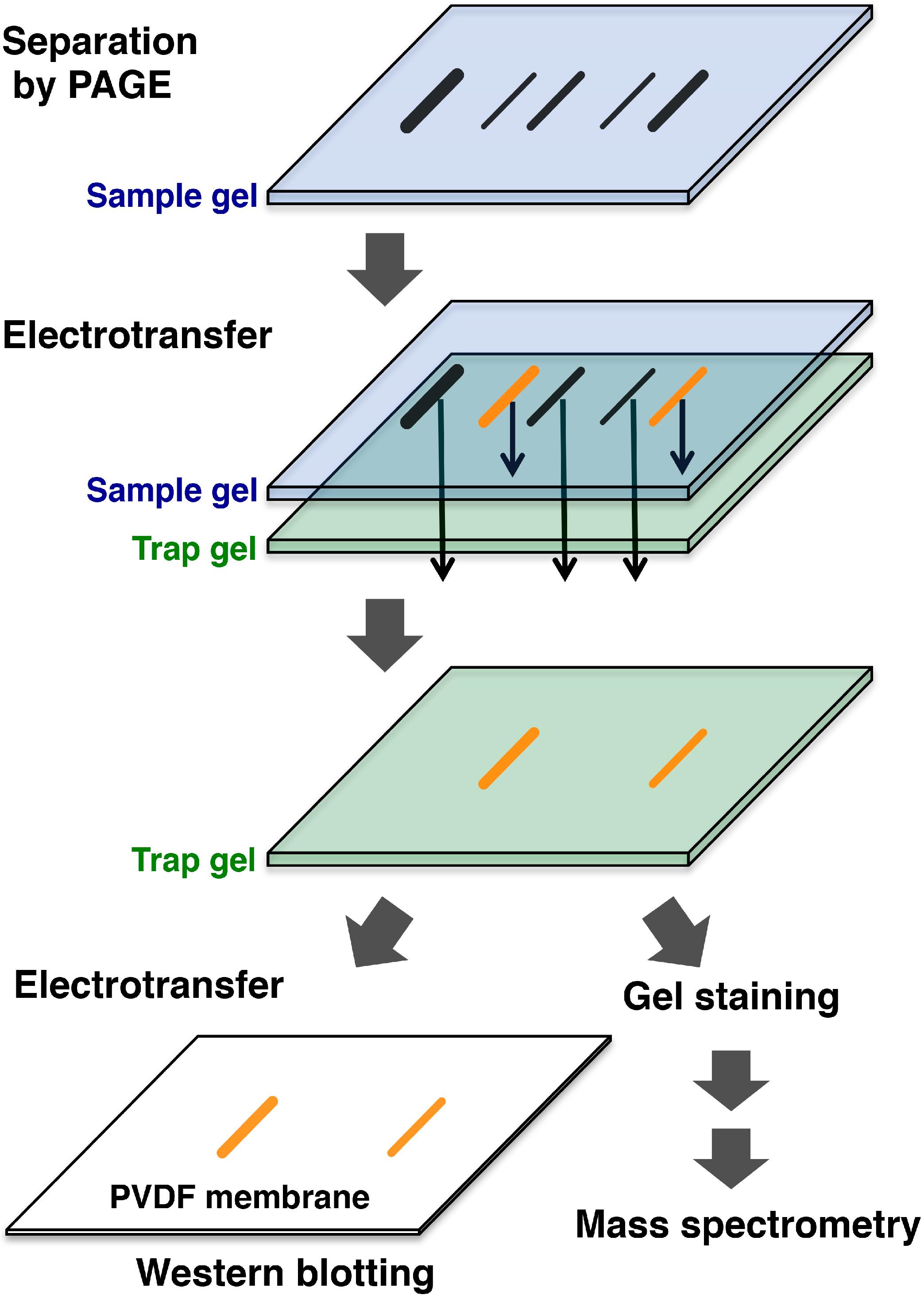
3. Affinity-Trap Polyacrylamide Gel Electrophoresis
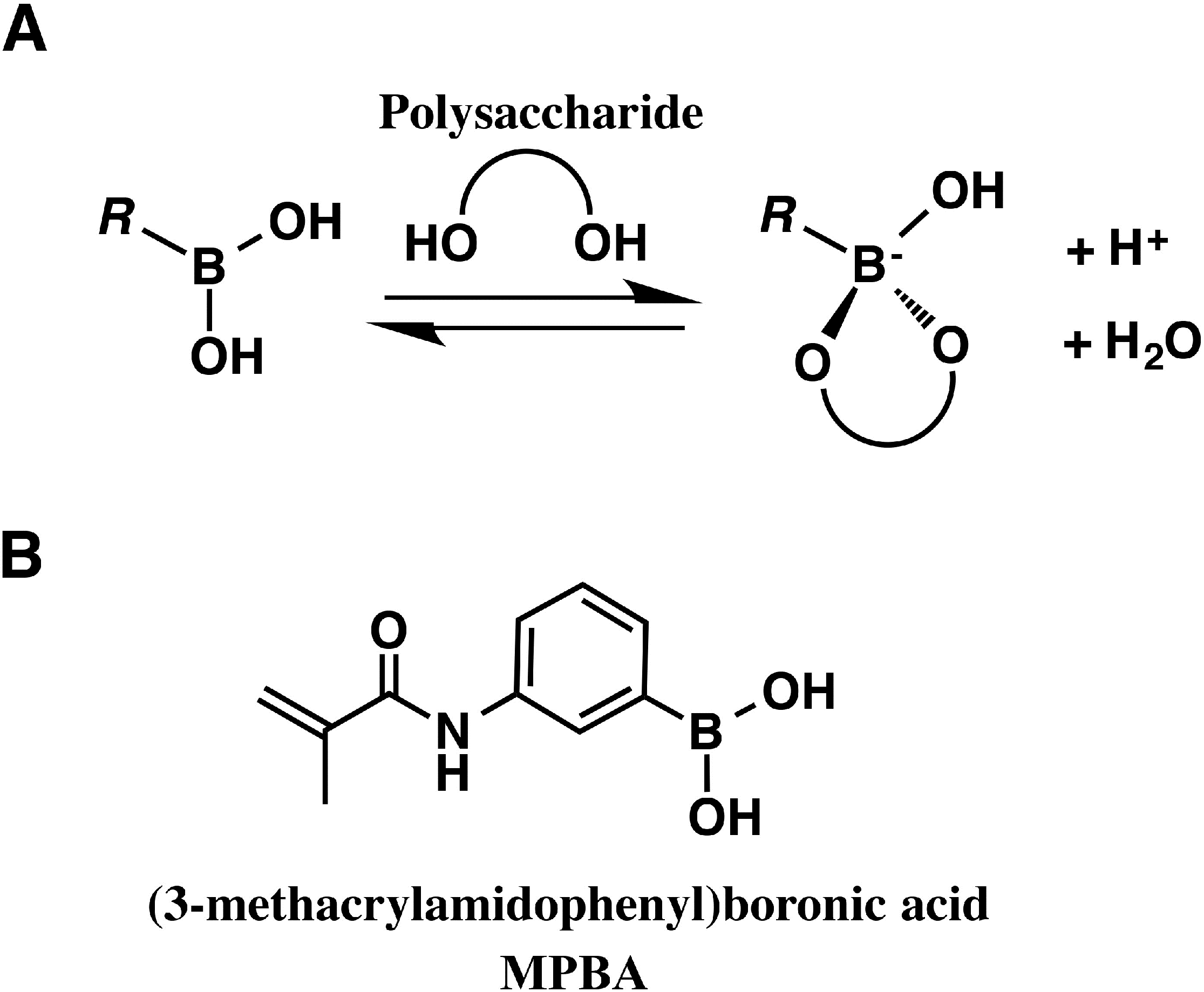
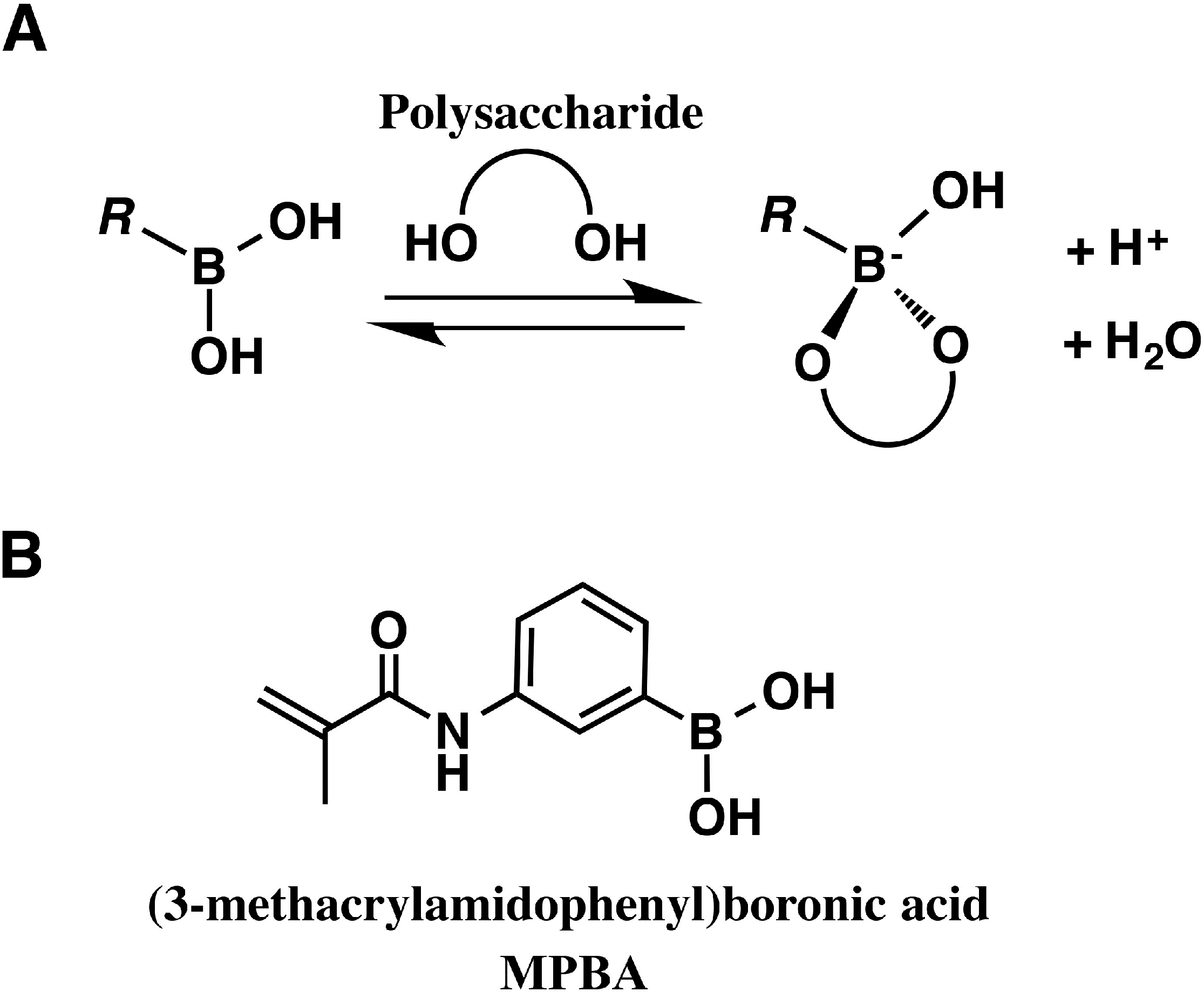
4. Saccharide Affinity Electrophoresis
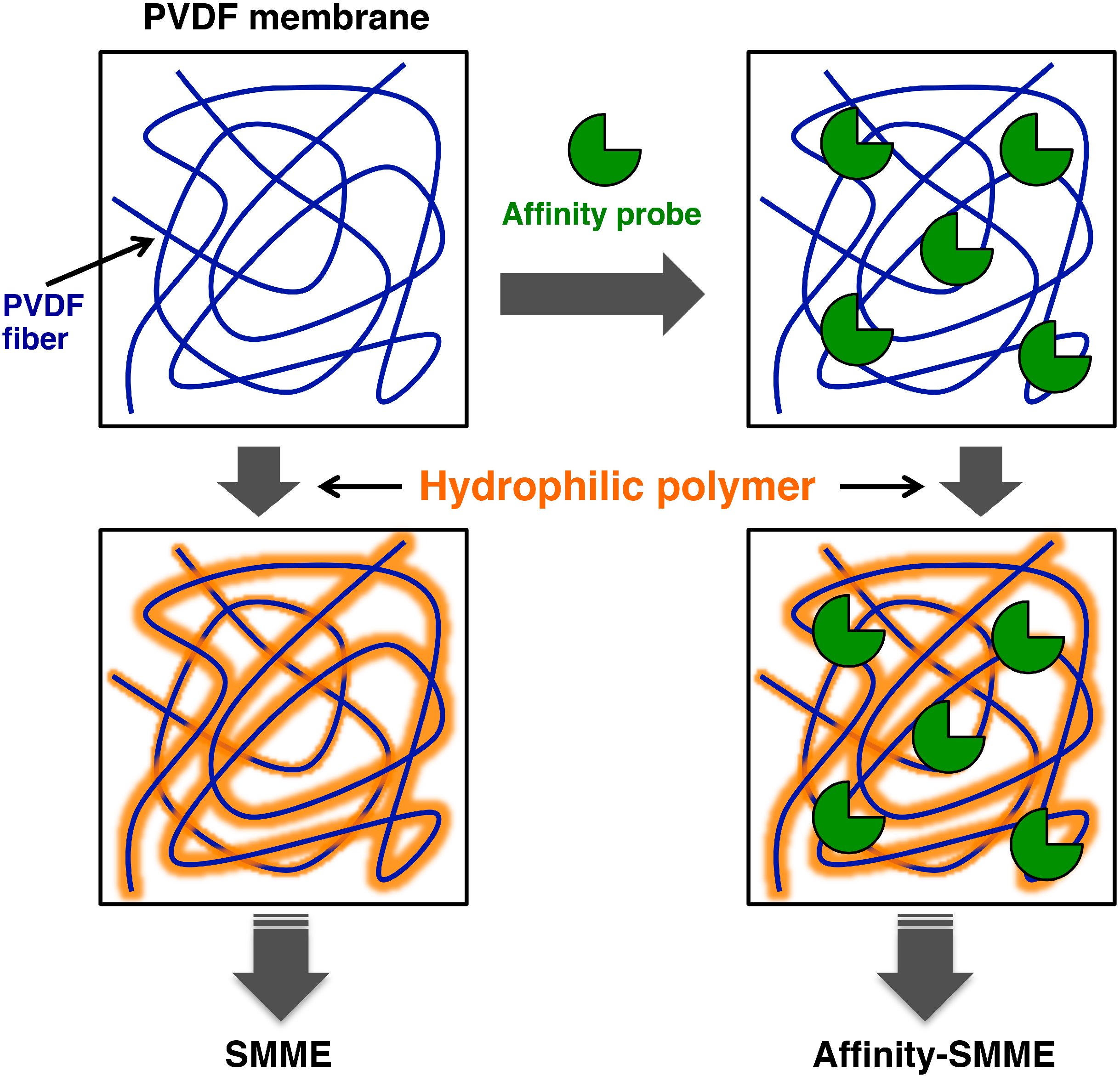
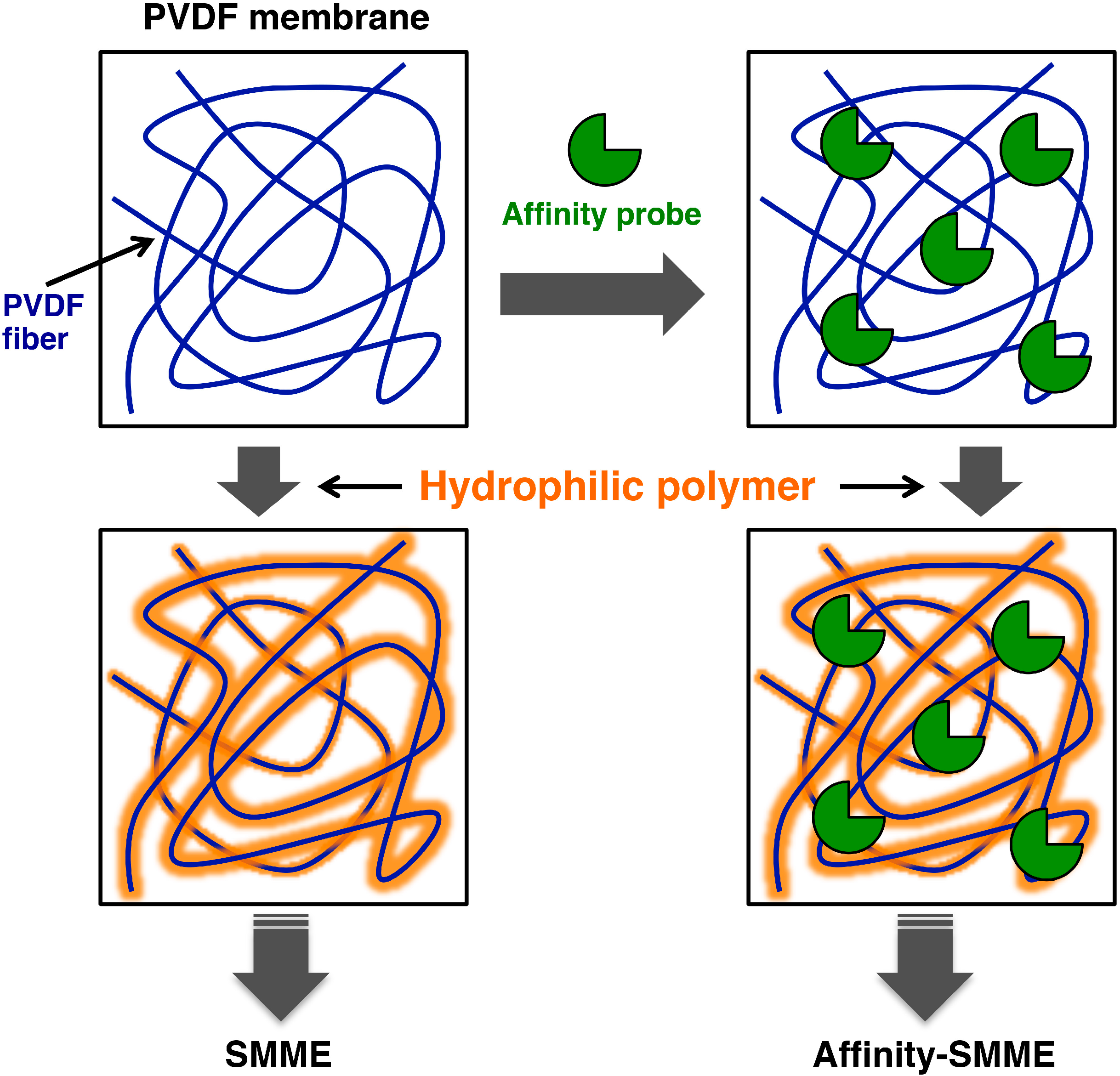
5. Supported Molecular Matrix Electrophoresis and Its Application to Affinity Electrophoresis
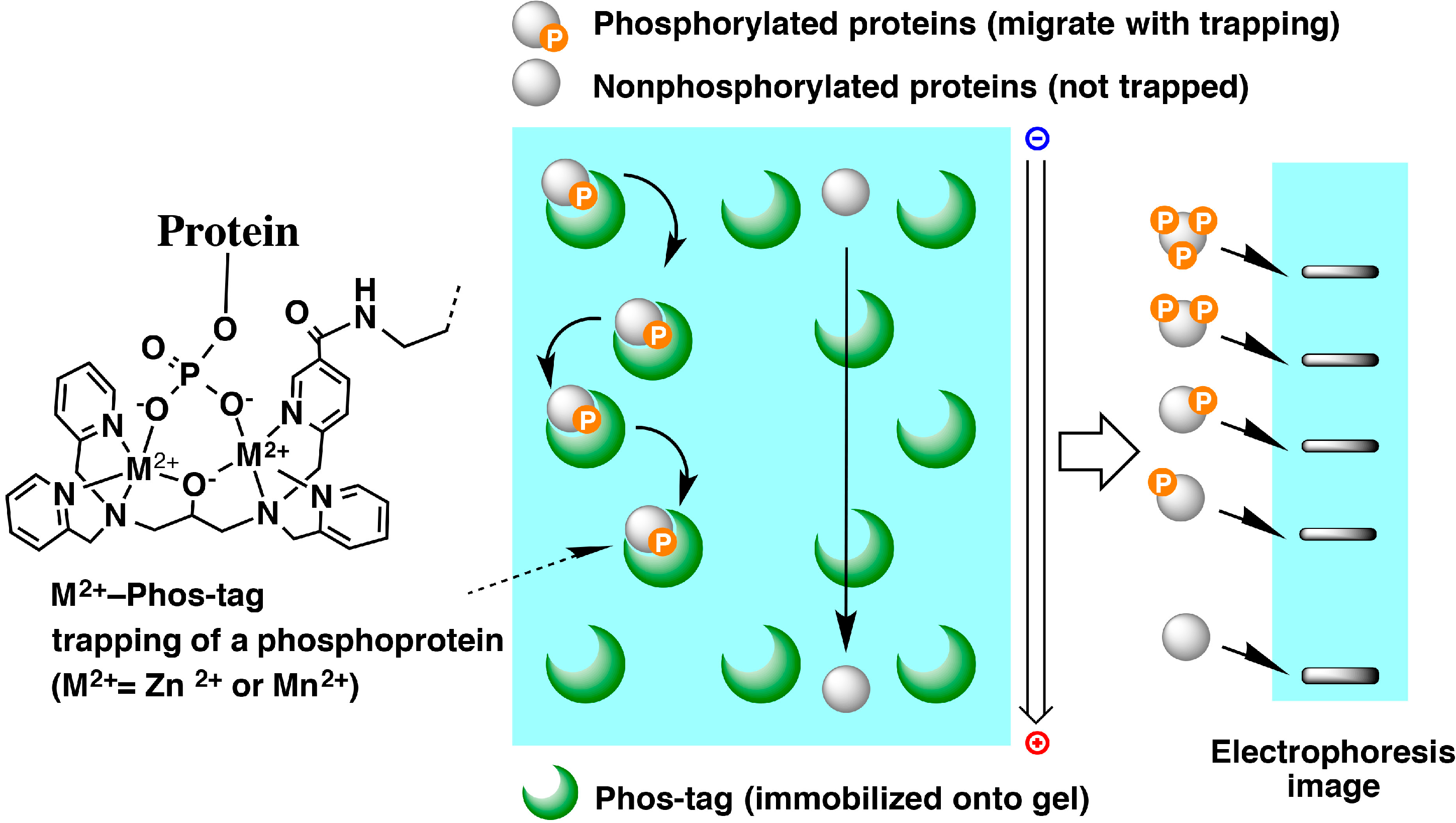
6. Phosphate Affinity Electrophoresis
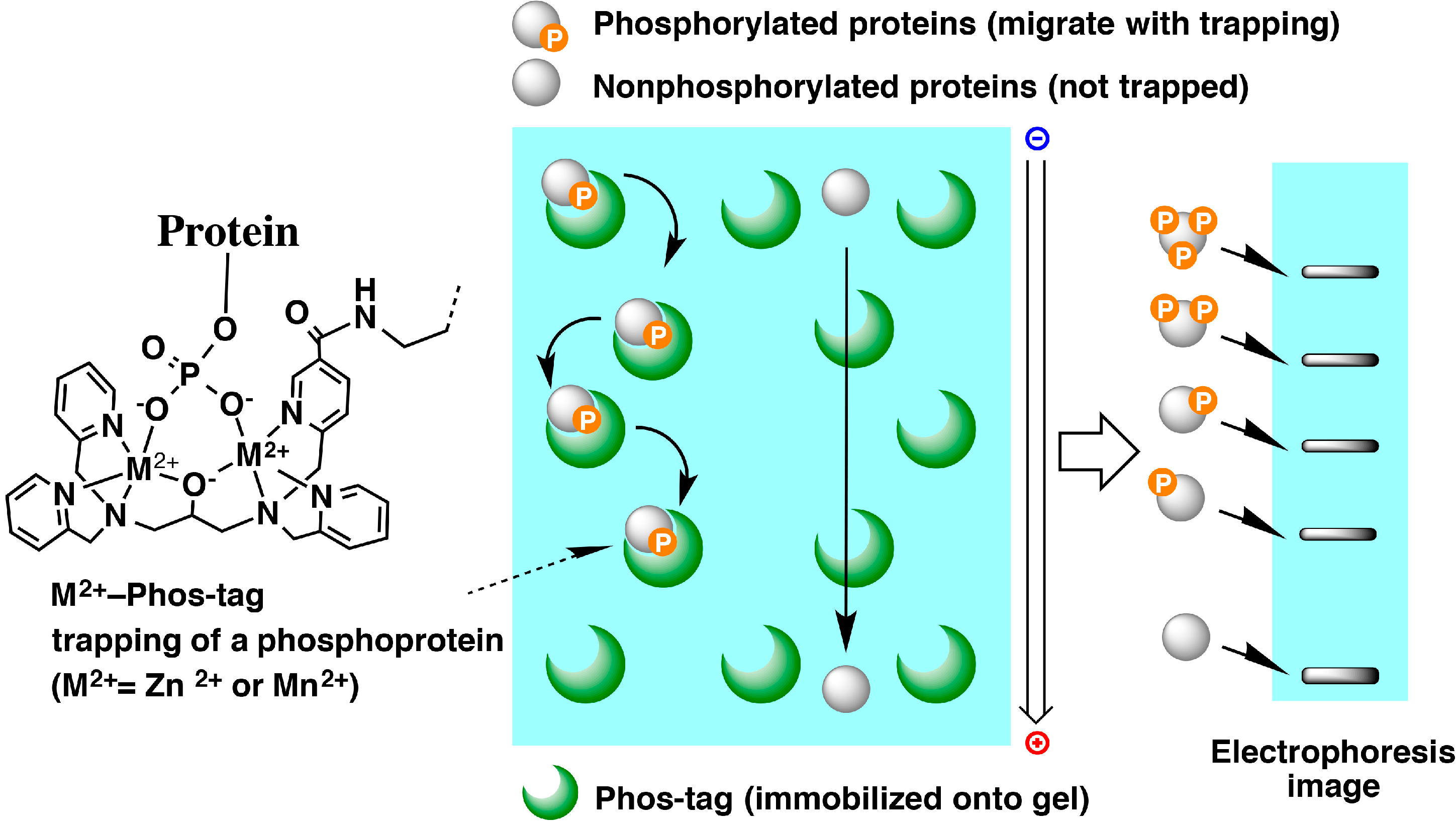
6.1. Examples of Analyses of Phosphorylated Proteins by Mn2+-Phos-Tag SDS-PAGE
6.2. Development of an Improved Protocol, Neutral Phos-Tag SDS-PAGE
6.3. Analysis of Chemically Unstable Histidine- or Aspartic Acid-Phosphorylated Proteins
6.4. Development of Microchip-Based Phos-Tag Electrophoresis
7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakamura, S.; Takeo, K.; Sasaki, I.; Murata, M. Proof of the formation of enzyme-substrate complex by “crossing-paper electrophoresis”. Nature 1959, 184, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takeo, K.; Tanaka, K.; Ueta, T. Crossing methods in paper electrophoresis. Hoppe-Seyler’s Z. Physiol. Chem. 1960, 318, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takeo, K.; Sasaki, I. Detection of enzyme substrate complexes by crossing electrophoresis. Hoppe-Seyler’s Z. Physiol. Chem. 1962, 328, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takeo, K.; Sasaki, I. Demonstration of enzyme substrate complexes in inactive or inactivated enzymes. Hoppe-Seyler’s Z. Physiol. Chem. 1963, 334, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Takeo, K.; Nakamura, S. Dissociation constants of glucan phosphorylases of rabbit tissues studied by polyacrylamide gel disc electrophoresis. Arch. Biochem. Biophys. 1972, 153, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Takeo, K.; Nitta, K.; Nakamura, S. Demonstration of phosphorylase in urines by polyacrylamide gel disc electrophoresis. Clin. Chim. Acta 1974, 57, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Takeo, K.; Kabat, E.A. Binding constants of dextrans and isomaltose oligosaccharides to dextran-specific myeloma proteins determined by affinity electrophoresis. J. Immunol. 1978, 121, 2305–2310. [Google Scholar] [PubMed]
- Sugii, S.; Takeo, K.; Kabat, E.A. Binding constants of NZB myeloma antidextrans for dextrans and isomaltose oligosaccharides determined by affinity electrophoresis. J. Immunol. 1979, 123, 1162–1168. [Google Scholar]
- Bøg-Hansen, T.C.; Takeo, K. Determination of dissociation constants by affinity electrophoresis: Complexes between human serum proteins and concanavalin A. Electrophoresis 1980, 1, 67–71. [Google Scholar] [CrossRef]
- Shimura, K. Progress in affinophoresis. J. Chromatogr. 1990, 510, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 2006, 5, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.R.; Springall, J.S.; Rogalle, D.; Masumoto, N.; Li, H.C.; D’Hooge, F.; Perera, S.P.; Jenkins, T.A.; James, T.D.; Fossey, J.S.; et al. Boronate affinity saccharide electrophoresis: A novel carbohydrate analysis tool. Electrophoresis 2008, 29, 4185–4191. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.P.P.; Mackay, J.D.; Bhamra, S.K.; Buchanan, J.G.; James, T.D.; Fossey, J.S.; van den Elsen, J.M.H. Analysis of protein glycation using phenylboronate acrylamide gel electrophoresis. Proteomics 2010, 10, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Aikyo, Y.; Oh-Ishi, M. Analysis of glycoproteins by SDS-PAGE with methacrylamido phenyl boronic acid. Electrophor. Lett. (Seibutsu Butsuri Kagaku) 2014, 58, 33–35. [Google Scholar] [CrossRef]
- Shimura, K.; Karger, B.L. Affinity probe capillary electrophoresis: Analysis of recombinant human growth hormone with a fluorescent labeled antibody fragment. Anal. Chem. 1994, 66, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shimura, K.; Kasai, K. Affinity probe capillary electrophoresis of insulin using a fluorescence-labeled recombinant Fab as an affinity probe. Electrophoresis 2014, 35, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; Wellner, E. Detection of cerebral spinal fluid-associated chemokines in birth traumatized premature babies by chip-based immunoaffinity CE. Electrophoresis 2013, 34, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Hosokawa, K.; Maeda, M. Phosphate-affinity electrophoresis on a microchip for determination of protein kinase activity. Electrophoresis 2009, 30, 3507–3513. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Hosokawa, K.; Maeda, M. Activity measurement of protein kinase and protein phosphatase by microchip phosphate-affinity electrophoresis. Anal. Biochem. 2012, 421, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Hosokawa, K.; Maeda, M. Application of microchip phosphate-affinity electrophoresis to measurement of protease activity in complex samples. Anal. Biochem. 2013, 432, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Krylov, S.N. Kinetic CE: Foundation for homogeneous kinetic affinity methods. Electrophoresis 2007, 28, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.-K.; Saito, T.; Gotoh, M.; Narimatsu, H.; Kameyama, A. Supported molecular matrix electrophoresis: A new tool for characterization of glycoproteins. Anal. Chem. 2009, 81, 3816–3823. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.-K.; Dong, W.; Yokoyama, S.; Yonezawa, S.; Saito, T.; Gotoh, M.; Narimatsu, H.; Kameyama, A. Improved method for immunostaining of mucin separated by supported molecular matrix electrophoresis by optimizing the matrix composition and fixation procedure. Electrophoresis 2011, 32, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.-K.; Dong, W.; Yokoyama, S.; Yonezawa, S.; Narimatsu, H.; Kameyama, A. Identification of mucins by using a method involving a combination of on-membrane chemical deglycosylation and immunostaining. J. Immunol. Methods 2013, 394, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.-K.; Kameyama, A. Affinity electrophoresis on supported molecular matrix electrophoresis. Electrophor. Lett. (Seibutsu Butsuri Kagaku) 2014, 58, 27–29. [Google Scholar] [CrossRef]
- Kinoshita-Kikuta, E.; Aoki, Y.; Kinoshita, E.; Koike, T. Label-free kinase profiling using phosphate affinity polyacrylamide gel electrophoresis. Mol. Cell. Proteomics 2007, 6, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Matsubara, M.; Aoki, Y.; Ohie, S.; Mouri, Y.; Koike, T. Two-dimensional phosphate-affinity gel electrophoresis for the analysis of phosphoprotein isotypes. Electrophoresis 2009, 30, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Ujihara, H.; Koike, T. Mobility shift detection of phosphorylation on large proteins using a Phos-tag SDS-PAGE gel strengthened with agarose. Proteomics 2009, 9, 4098–4101. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E. Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 2011, 11, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Phos-tag SDS-PAGE systems for phosphorylation profiling of proteins with a wide range of molecular masses under neutral pH conditions. Proteomics 2012, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Kikuta, E.; Kinoshita, E.; Koike, T. Separation and identification of four distinct serine-phosphorylation states of ovalbumin by Phos-tag affinity electrophoresis. Electrophoresis 2012, 33, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Kikuta, E.; Kinoshita, E.; Koike, T. A laborsaving, timesaving, and more reliable strategy for separation of low-molecular-mass phosphoproteins in Phos-tag affinity electrophoresis. Int. J. Chem. 2012, 4, 1–8. [Google Scholar] [CrossRef]
- Awada, C.; Sato, T.; Takao, T. Affinity-trap polyacrylamide gel electrophoresis: A novel method of capturing specific proteins by electro-transfer. Anal. Chem. 2010, 82, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Oh-Ishi, M.; Satoh, M.; Maeda, T. Preparative two-dimensional gel electrophoresis with agarose gels in the first dimension for high molecular mass proteins. Electrophoresis 2000, 21, 1653–1669. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Takahashi, M.; Takeda, H.; Shiro, M.; Koike, T. Recognition of phosphate monoester dianion by an alkoxide-bridged dinuclear zinc(II) complex. Dalton Trans. 2004, 21, 1189–1193. [Google Scholar] [CrossRef]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Matsubara, M.; Yamada, S.; Nakamura, H.; Shiro, Y.; Aoki, Y.; Okita, K.; Koike, T. Separation of phosphoprotein isotypes having the same number of phosphate groups using phosphate-affinity SDS-PAGE. Proteomics 2008, 8, 2994–3003. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Phosphate-affinity gel electrophoresis using a Phos-tag molecule for phosphoproteome study. Curr. Proteomics 2009, 6, 104–121. [Google Scholar] [CrossRef]
- Kimura, Y.; Nagata, K.; Suzuki, N.; Yokoyama, R.; Yamanaka, Y.; Kitamura, H.; Hirano, H.; Ohara, O. Characterization of multiple alternative forms of heterogeneous nuclear ribonucleoprotein K by phosphate-affinity electrophoresis. Proteomics 2010, 10, 3884–3895. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Saito, T.; Asada, A.; Fukunaga, K.; Hisanaga, S. Quantitative measurement of in vivo phosphorylation states of Cdk5 activator p35 by Phos-tag SDS-PAGE. Mol. Cell. Proteomics 2010, 9, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Takeya, K.; Loutzenhiser, K.; Shiraishi, M.; Loutzenhiser, R.; Walsh, M.P. A highly sensitive technique to measure myosin regulatory light chain phosphorylation: The first quantification in renal arterioles. Am. J. Physiol. Ren. Physiol. 2008, 294, F1487–F1492. [Google Scholar] [CrossRef]
- El-Yazbi, A.F.; Johnson, R.P.; Walsh, E.J.; Takeya, K.; Walsh, M.P.; Cole, W.C. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J. Physiol. 2010, 588, 1747–1762. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.N.; Tracey, C.N.; Tsang, S.C.; McGinnis, J.M.; Mitchell, B.F. Phos-tag-based analysis of myosin regulatory light chain phosphorylation in human uterine myocytes. PLoS One 2011, 6, e20903. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirano, M.; Ide, T.; Ichiki, T.; Koibuchi, N.; Sunagawa, K.; Hirano, K. Pivotal role of Rho-associated kinase 2 in generating the intrinsic circadian rhythm of vascular contractility. Circulation 2013, 127, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nakamura, H.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; Shiro, Y. Separation of a phosphorylated histidine protein using phosphate affinity polyacrylamide gel electrophoresis. Anal. Biochem. 2007, 360, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Shiba, A.; Edahiro, K.; Inoue, Y.; Yamamoto, K.; Yoshida, M.; Koike, T. Profiling of protein thiophosphorylation by Phos-tag affinity electrophoresis: Evaluation of adenosine 5'-O-(3-thiotriphosphate) as a phosphoryl donor in protein kinase reactions. Proteomics 2014, 14, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Wayne, K.J.; Li, S.; Kazmierczak, K.M.; Tsui, H.C.; Winkler, M.E. Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Mol. Microbiol. 2012, 86, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, A.; Chen, Q.; Hinton, D.M.; Stibitz, S. In vivo phosphorylation dynamics of the Bordetella pertussis virulence-controlling response regulator BvgA. Mol. Microbiol. 2013, 88, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Kaimer, C.; Zusman, D.R. Phosphorylation-dependent localization of the response regulator FrzZ signals cell reversals in Myxococcus xanthus. Mol. Microbiol. 2013, 88, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Ishii, E.; Eguchi, Y.; Utsumi, R. Mechanism of activation of PhoQ/PhoP two-component signal transduction by SafA, an auxiliary protein of PhoQ histidine kinase in Escherichia coli. Biosci. Biotechnol. Biochem. 2013, 77, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Li, R.F.; Lu, G.T.; Li, L.; Su, H.Z.; Feng, G.F.; Chen, Y.; He, Y.Q.; Jiang, B.L.; Tang, D.J.; Tang, J.L.; et al. Identification of a putative cognate sensor kinase for the two-component response regulator HrpG, a key regulator controlling the expression of the hrp genes in Xanthomonas campestris pv. campestris. Environ. Microbiol. 2014, 16, 2053–2071. [Google Scholar] [CrossRef]
- Madec, E.; Bontemps-Gallo, S.; Lacroix, J.M. Increased phosphorylation of the RcsB regulator of the RcsCDB phosphorelay in strains of Dickeya dadantii devoid of osmoregulated periplasmic glucans revealed by Phos-tag gel analysis. Microbiology 2014, 160, 2763–2770. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.J.; Li, M.; Brinkworth, C.S.; Paulson, J.L.; Wang, D.; Hübner, A.; Chou, W.-H.; Davis, R.J.; Burlingame, A.L.; Messing, R.O.; et al. A semisynthetic epitope for kinase substrates. Nat. Methods 2007, 4, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kee, J.-M.; Muir, T.W. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem. Biol. 2012, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.K.; Plate, L.; Price, M.S.; Allen, J.J.; Shokat, K.M. Use of a semisynthetic epitope to probe histidine kinase activity and regulation. Anal. Biochem. 2010, 397, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Wilke, K.E.; Francis, S.; Carlson, E.E. Activity-based probe for histidine kinase signaling. J. Am. Chem. Soc. 2012, 134, 9150–9153. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. The Cutting Edge of Affinity Electrophoresis Technology. Proteomes 2015, 3, 42-55. https://doi.org/10.3390/proteomes3010042
Kinoshita E, Kinoshita-Kikuta E, Koike T. The Cutting Edge of Affinity Electrophoresis Technology. Proteomes. 2015; 3(1):42-55. https://doi.org/10.3390/proteomes3010042
Chicago/Turabian StyleKinoshita, Eiji, Emiko Kinoshita-Kikuta, and Tohru Koike. 2015. "The Cutting Edge of Affinity Electrophoresis Technology" Proteomes 3, no. 1: 42-55. https://doi.org/10.3390/proteomes3010042
APA StyleKinoshita, E., Kinoshita-Kikuta, E., & Koike, T. (2015). The Cutting Edge of Affinity Electrophoresis Technology. Proteomes, 3(1), 42-55. https://doi.org/10.3390/proteomes3010042