Ursolic Acid-Regulated Energy Metabolism—Reliever or Propeller of Ultraviolet-Induced Oxidative Stress and DNA Damage?

Abstract
:1. Background
1.1. Effects of UV in Sunlight
1.2. Ursolic Acid and Its Biological Functions
1.2.1. Antioxidant Activity
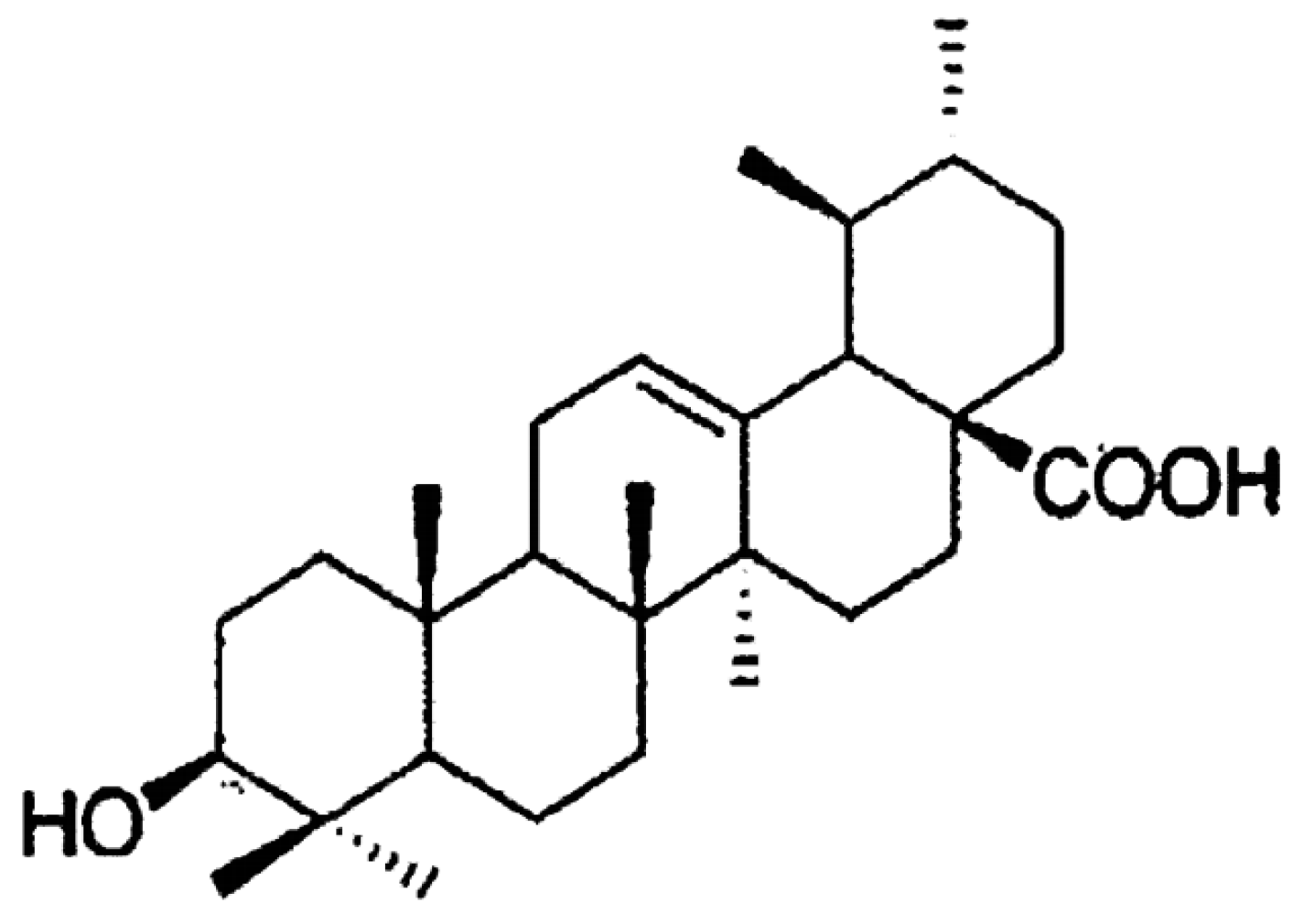
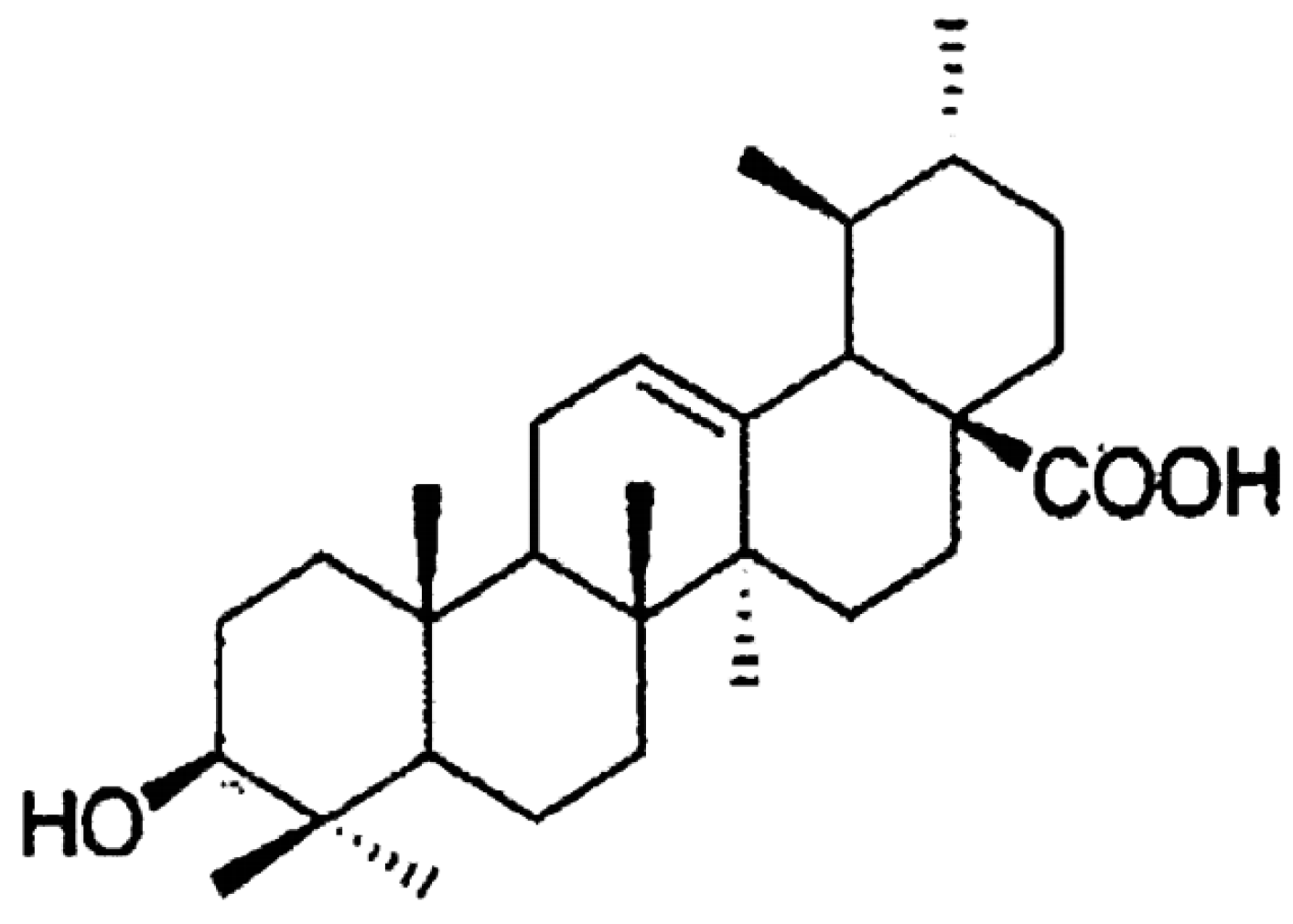
1.2.2. Capabilities of Ceramide Stabilization and Surface Protein Recognition
1.2.3. Antitumoral Activity
2. UV-Induced Mitogenic Activation for Cell Survival
2.1. UV-Induced Adaptive Defense against TKR-Mediated Mitogenic Effect
2.2. UV-induced Cell Lethality through TKR-mediated Mitogenic Effects
3. Kinase Activation by UA Modulates UV-Induced Oxidative DNA Damage
3.1. UA-Induced p53 Activation and Modulation of UV-Invoked ROS
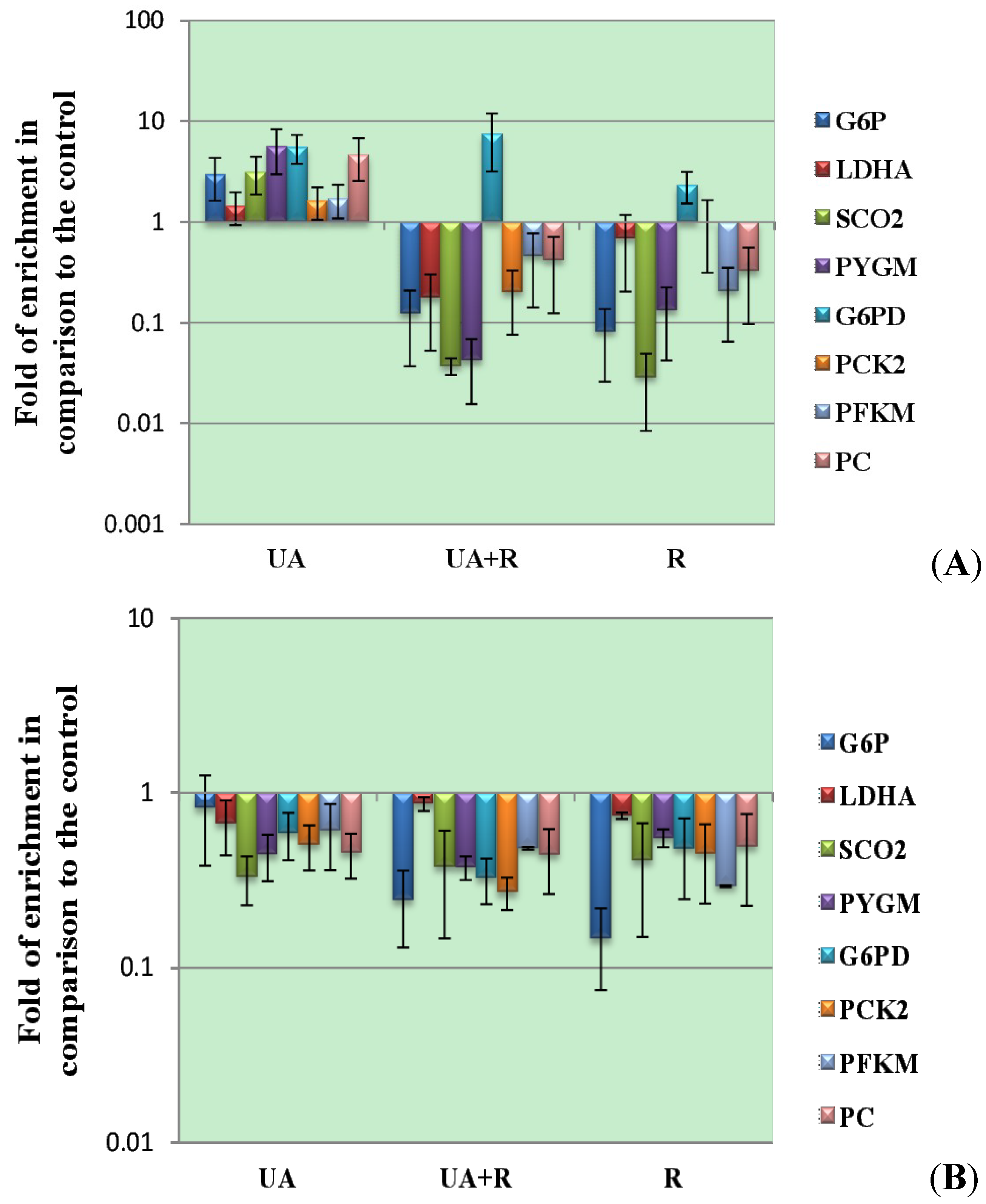
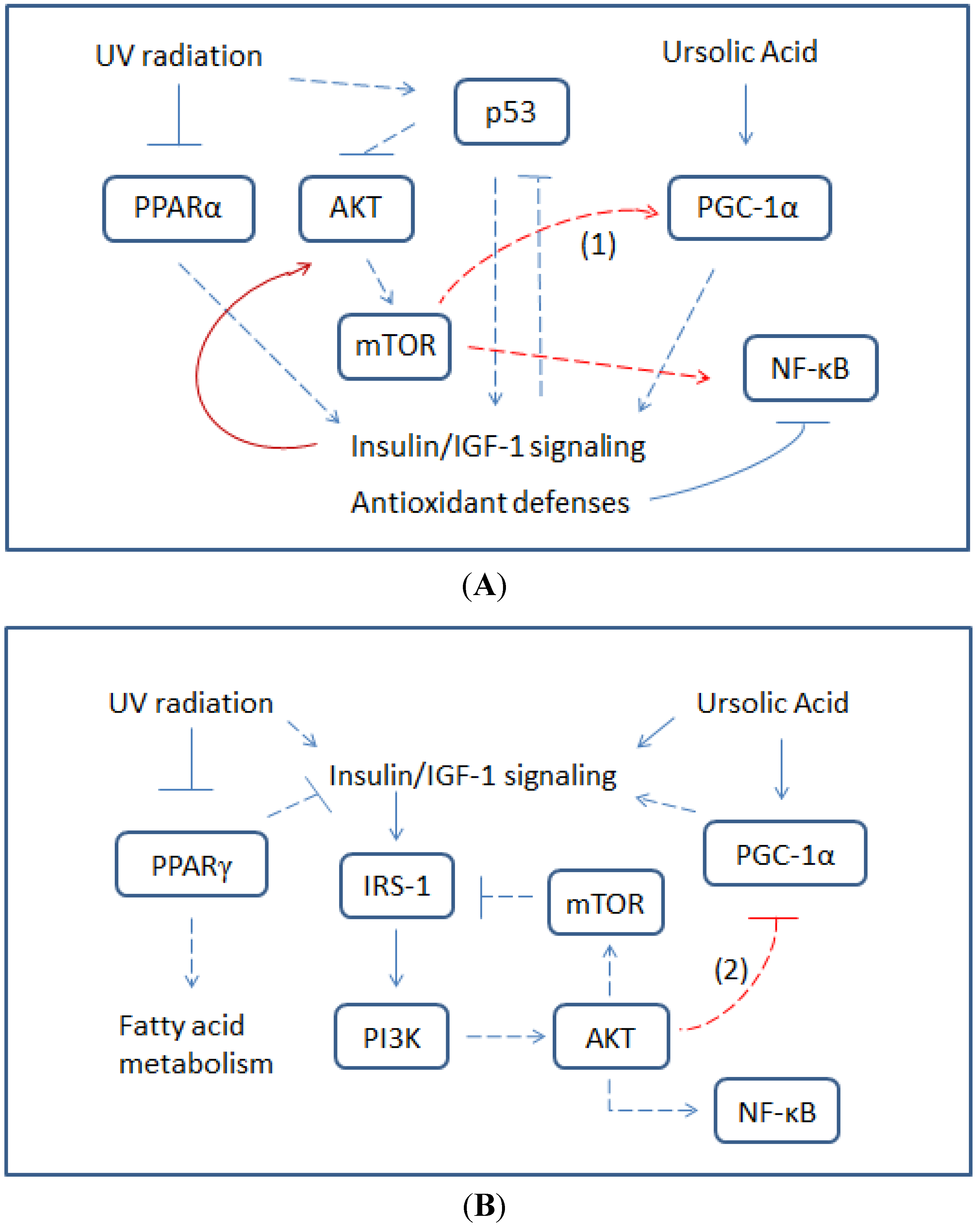
3.2. Modulatory Effects of UA on Cellular Response to UV-Induced TKR Down-Regulation

{kind=link}
{kind=link}
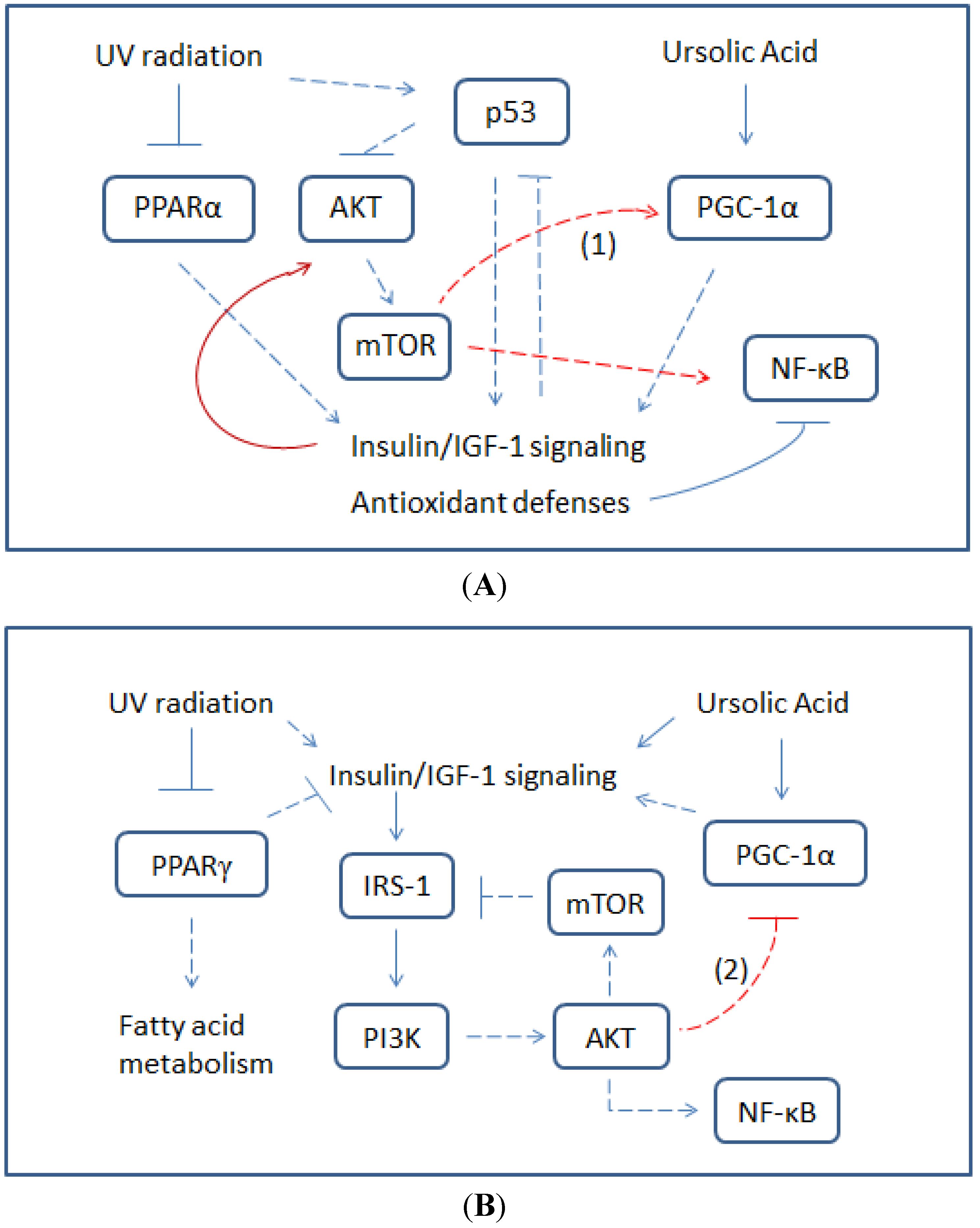{kind=link}
{kind=link}
| Enzymes | mRNA Expression | Cell Lines, Tissues or Organs Invetigated |
|---|---|---|
| Glucose-6-phosphatase | Up-regulation | Human hepatic carcinoma and mouse liver [127,129] |
| Phosphoenolpyruvate carboxykinase | Up-regulation | Human hepatic carcinoma [129] |
| Phosphofructokinase | Down-regulation | Mouse skeletal muscle [128] |
| Lactate dehydrogenase A | Down-regulation | Mouse skeletal muscle [127] |
| Cytochrome c oxidase | Up-regulation | Human kidney [128] |
| Glycogen phosphorylase | Down-regulation | Mouse skeletal muscle [128] |
4. Conclusions
Abbreviations
| ATM | ataxia telangiectasia mutated |
| AMPK | AMP-activated protein kinase |
| cAMP | cyclic adenosine monophosphate |
| CPD | cyclobutane pyrimidine dimers |
| DNA-PK | DNA-dependent protein kinase |
| G6PD | glucose-6-phosphate dehydrogenase |
| GSK3β | glycogen synthase kinase 3β |
| HRR | homologous recombination repair |
| IGF-1 | insulin-like growth factor 1 |
| IRS-1 | insulin receptor substrate 1 |
| MAPK | mitogen-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHEJ | non-homologous end-joining |
| Nrf2 | nuclear factor erythroid-derived 2-related factor 2 |
| PGC-1α | PPAR-γ coactivator 1α |
| PIKK | phosphatidylinositol 3-kinase-related kinase |
| PKA | cAMP-dependent protein kinase |
| PPAR | peroxisome proliferator-activated receptor |
| PPP | pentose phosphate pathway |
| redox | reduction-oxidation |
| ROS | reactive oxygen species |
| RPE | retinal pigment epithelium |
| TKR | tyrosine kinase-coupled receptor |
| UA | ursolic acid |
| UV | ultraviolet |
| UV-VIS | (or UVR) radiation with spectrum wavelength ranging from UV to visible light |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Geography 474: Energy Interactions with the Atmosphere and at the Surface. Department of Geography, University of Delaware. Available online: http://www.udel.edu/Geography/DeLiberty/Geog474/geog474_energy_interact.html (accessed on 3 July 2013).
- CHAPTER 3 Energy Sources, Transfers and Transformations: Terrestrial Radiation. Available online: http://atlantic.evsc.virginia.edu/~bph/AW_Book_Spring_96/AW_Book_29.html (accessed on 3 July 2013).
- Ali, S.M.; Bonnier, F.; Ptasinski, K.; Lambkin, H.; Flynn, K.; Lyng, F.M.; Byrne, H.J. Raman spectroscopic mapping for the analysis of solar radiation induced skin damage. Analyst 2013, 138, 3946–3956. [Google Scholar] [CrossRef]
- Light Source Spectrum Wavelength Ranges. Lumen Dynamics. Available online: http://www.ldgi.com/technology-learning-center/uv-light/typical-light-source-spectrum/ (accessed on 29 May 2013).
- Françoise, L. (Ed.) Société de chimie physique. In Photophysics and Photochemistry above 6 eV, Proceedings of the 38th International Meeting of the Société de chimie physique, Bombannes, France, 17–21 September 1984; Elsevier Science Pub. Co.: New York, NY, USA, 1985.
- Niemz, M.H. Laser-Tissue Interactions: Fundamentals and Applications, 2nd ed.; Springer: Berlin, Germany, 2002; pp. 45–58. [Google Scholar]
- Love, J.D.; Nguyen, H.T.; Or, A.; Attri, A.K.; Minton, K.W. UV-Induced interstrand cross-linking of d(GT)n.d(CA)n is facilitated by a structural transition. J. Biol. Chem. 1986, 261, 10051–10057. [Google Scholar]
- Deby, C.; Goutier, R. New Perspectives on the Biochemistry of Superoxide Anion and the Efficiency of Superoxide Dismutases. Biochem. Pharmacol. 1990, 39, 399–405. [Google Scholar] [CrossRef]
- Fernandez, T.L.; van Lonkhuyzen, D.R.; Dawson, R.; Kimlin, M.G.; Upton, Z. Characterisation of a human skin equivalent model to study the effects of ultraviolet B radiation on keratinocytes. Tissue Eng. Part C Methods 2013, 20, 588–598. [Google Scholar]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar] [CrossRef]
- Rünger, T.M. C→T Transition mutations are not solely UVB-signature mutations, because they are also generated by UVA. J. Invest. Dermatol. 2008, 128, 2138–2140. [Google Scholar] [CrossRef]
- Schauen, M.; Hornig-Do, H.-T.; Schomberg, S.; Herrmann, G.; Wiesner, R.J. Mitochondrial electron transport chain activity is not involved in ultraviolet A (UVA)-induced cell death. Free Radic. Biol. Med. 2007, 42, 499–509. [Google Scholar] [CrossRef]
- Trojanek, J.; Ho, T.; del Valle, L.; Nowicki, M.; Wang, J.Y.; Lassak, A.; Peruzzi, F.; Khalili, K.; Skorski, T.; Reiss, K. Role of the insulin-like growth factor I/Insulin receptor substrate 1 axis in Rad51 trafficking and DNA repair by homologous recombination. Mol. Cell. Biol. 2003, 23, 7510–7524. [Google Scholar] [CrossRef]
- Kuhn, C.; Hurwitz, S.A.; Kumar, M.G.; Cotton, J.; Spandau, D.F. Activation of the insulin-like growth factor-1 receptor promotes the survival of human keratinocytes following ultraviolet B irradiation. Int. J. Cancer 1999, 80, 431–438. [Google Scholar] [CrossRef]
- Rajala, R.V.S.; Anderson, R.E. Light regulation of the insulin receptor in the retina. Mol. Neurobiol. 2003, 28, 123–138. [Google Scholar] [CrossRef]
- Bell, M.W.; Alvarez, K.; Ghalayini, A.J. Association of the tyrosine phosphatase SHP-2 with transducin-alpha and a 97-kDa tyrosine-phosphorylated protein in photoreceptor rod outer segments. J. Neurochem. 1999, 73, 2331–2340. [Google Scholar]
- Ghalayini, A.J.; Guo, X.X.; Koutz, C.A.; Anderson, R.E. Light stimulates tyrosine phosphorylation of rat rod outer segments in vivo. Exp. Eye Res. 1998, 66, 817–821. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Wang, E.; Kumar, N.; Glickman, R.D. Ursolic acid differentially modulates apoptosis in skin melanoma and retinal pigment epithelial cells exposed to UV-VIS broadband radiation. Apoptosis 2013, 19, 861–828. [Google Scholar]
- Phytochemicals Health Promotion and Therapeutic Potential; Carkeet, C.; Grann, K.; Randolph, R.K.; Venzon, D.S.; Izzy, S. (Eds.) Taylor & Francis/CRC Press: Boca Raton, FL, USA, 2013; pp. 120–128.
- Martin-Aragón, S.; de las Heras, B.; Sanchez-Reus, M.I.; Benedi, J. Pharmacological modification of endogenous antioxidant enzymes by ursolic acid on tetrachloride-induced liver damage in rats and primary cultures of rat hepatocytes. Exp. Toxicol. Pathol. 2001, 53, 199–206. [Google Scholar] [CrossRef]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral role of noxa in p53-mediated apoptotic response. Genes Dev. 2003, 17, 2233–2238. [Google Scholar] [CrossRef]
- Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–68. [Google Scholar] [CrossRef]
- Ramachandran, S.; Prasad, N.R. Effect of ursolic acid, a triterpenoid antioxidant, on ultraviolet-B radiation-induced cytotoxicity, lipid peroxidation and DNA damage in human lymphocytes. Chem. Biol. Interact. 2008, 176, 99–107. [Google Scholar] [CrossRef]
- Liobikas, J.; Majiene, D.; Trumbeckaite, S.; Kursvietiene, L.; Masteikova, R.; Kopustinskiene, D.M.; Savickas, A.; Bernatoniene, J. Uncoupling and antioxidant effects of ursolic acid in isolated rat heart mitochondria. J. Nat. Prod. 2011, 74, 1640–1644. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kumar, N.C.; Glickman, R.D. Modulation of photochemical damage in normal and malignant cells by naturally occurring compounds. Photochem. Photobiol. 2012, 88, 1385–1395. [Google Scholar]
- Chen, K.-C.; Chang, H.-H.; Ko, W.-S.; Wu, C.-L.; Chiu, W.-T.; Hsieh, C.-L.; Peng, R.Y. UV-induced damages eliminated by arbutin and ursolic acid in cell model of human dermal fibroblast WS-1 cells. Egypt. Dermatol. Online J. 2009, 5, 1–15. [Google Scholar]
- Soo Lee, Y.; Jin, D.-Q.; Beak, S.-M.; Lee, E.-S.; Kim, J.-A. Inhibition of ultraviolet-A-modulated signaling pathways by asiatic acid and ursolic acid in HaCaT human keratinocytes. Eur. J. Pharmacol. 2003, 476, 173–178. [Google Scholar] [CrossRef]
- Bayer, M.; Proksch, P.; Felsner, I.; Brenden, H.; Kohne, Z.; Walli, R.; Duong, T.N.; Götz, C.; Krutmann, J.; Grether-Beck, S. Photoprotection against UVAR: Effective triterpenoids require a lipid raft stabilizing chemical structure. Exp. Dermatol. 2011, 20, 955–958. [Google Scholar] [CrossRef]
- Wilkinson, K.; Boyd, J.D.; Glicksman, M.; Moore, K.J.; El Khoury, J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J. Biol. Chem. 2011, 286, 34914–34922. [Google Scholar]
- Handberg, A.; Lopez-Bermejo, A.; Bassols, J.; Vendrell, J.; Ricart, W.; Fernandez-Real, J.M. Circulating soluble CD36 is associated with glucose metabolism and interleukin-6 in glucose-intolerant men. Diabetes Vasc. Dis. Res. 2009, 6, 15–20. [Google Scholar] [CrossRef]
- Samovski, D.; Su, X.; Xu, Y.; Abumrad, N.A.; Stahl, P.D. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J. Lipid Res. 2012, 53, 709–717. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Es-saady, D.; Simon, A.; Ollier, M.; Maurizis, J.C.; Chulia, A.J.; Delage, C. Inhibitory effect of ursolic acid on B16 proliferation through cell cycle arrest. Cancer Lett. 1996, 106, 193–197. [Google Scholar] [CrossRef]
- Tang, C.; Lu, Y.-H.; Xie, J.-H.; Wang, F.; Zou, J.-N.; Yang, J.-S.; Xing, Y.-Y.; Xi, T. Downregulation of survivin and activation of caspase-3 through the PI3K/Akt pathway in ursolic acid-induced HepG2 cell apoptosis. Anticancer Drugs 2009, 20, 249–258. [Google Scholar] [CrossRef]
- Yeh, C.-T.; Wu, C.-H.; Yen, G.-C. Ursolic acid, a naturally occurring triterpenoid, suppresses migration and invasion of human breast cancer cells by modulating c-Jun N-terminal kinase, Akt and mammalian target of rapamycin signaling. Mol.Nutr.Food Res. 2010, 54, 1285–1295. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.; Zheng, Y.; Hu, B.; Cheng, W.; Zhang, Z.; Shan, Q. Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IκB kinase Β/nuclear factor-κB-mediated inflammatory pathways in mice. Brain Behav. Immun. 2011, 25, 1658–1667. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef]
- Persson, C.; Sjöblom, T.; Groen, A.; Kappert, K.; Engström, U.; Hellman, U.; Heldin, C.-H.; den Hertog, J.; Östman, A. Preferential oxidation of the second phosphatase domain of receptor-like PTP-α revealed by an antibody against oxidized protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 2004, 101, 1886–1891. [Google Scholar] [CrossRef]
- Groß, S.; Knebel, A.; Tenev, T.; Neininger, A.; Gaestel, M.; Herrlich, P.; Böhmer, F.D. Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J. Biol. Chem. 1999, 274, 26378–26386. [Google Scholar]
- Khanna, K.K.; Shiloh, Y. The DNA Damage Response: Implications on Cancer Formation and Treatment: Implications on Cancer Formation and Treatment; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Dobbs, T.A.; Tainer, J.A.; Lees-Miller, S.P. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 2010, 9, 1307–1314. [Google Scholar] [CrossRef]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 Modulates Homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252. [Google Scholar] [CrossRef]
- Héron-Milhavet, L.; LeRoith, D. Insulin-like growth factor I induces MDM2-dependent degradation of p53 via the p38 MAPK pathway in response to DNA damage. J. Biol. Chem. 2002, 277, 15600–15606. [Google Scholar] [CrossRef]
- Héron-Milhavet, L.; Karas, M.; Goldsmith, C.M.; Baum, B.J.; LeRoith, D. Insulin-like growth factor-I (IGF-I) receptor activation rescues UV-damaged cells through a p38 signaling pathway. Potential role of the IGF-I receptor in DNA repair. J. Biol. Chem. 2001, 276, 18185–18192. [Google Scholar]
- Tao, W.; Levine, A.J. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef]
- Shaw, L.M. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the Alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol. 2001, 21, 5082–5093. [Google Scholar] [CrossRef]
- Pirola, L.; Bonnafous, S.; Johnston, A.M.; Chaussade, C.; Portis, F.; van Obberghen, E. Phosphoinositide 3-kinase-mediated reduction of insulin receptor substrate-1/2 protein expression via different mechanisms contributes to the insulin-induced desensitization of its signaling pathways in L6 muscle cells. J. Biol. Chem. 2003, 278, 15641–15651. [Google Scholar]
- Coffer, P.J.; Burgering, B.M.; Peppelenbosch, M.P.; Bos, J.L.; Kruijer, W. UV activation of receptor tyrosine kinase activity. Oncogene 1995, 11, 561–569. [Google Scholar]
- Thakur, S.; Garg, N.; Adamo, M.L. Deficiency of Insulin-like growth factor-1 receptor confers resistance to oxidative stress in C2C12 myoblasts. PLoS One 2013, 8, e63838. [Google Scholar] [CrossRef]
- Sertznig, P.; Reichrath, J. Peroxisome proliferator-activated receptors (PPARs) in dermatology. Dermato-Endocrinol. 2011, 3, 130–135. [Google Scholar]
- El Azzouzi, H.; Leptidis, S.; Bourajjaj, M.; Armand, A.-S.; van der Nagel, R.; van Bilsen, M.; da Costa Martins, P.A.; de Windt, L.J. Peroxisome proliferator-activated receptor (PPAR) gene profiling uncovers insulin-like growth factor-1 as a PPARalpha target gene in cardioprotection. J. Biol. Chem. 2011, 286, 14598–14607. [Google Scholar] [CrossRef]
- Zhang, Q.; Southall, M.D.; Mezsick, S.M.; Johnson, C.; Murphy, R.C.; Konger, R.L.; Travers, J.B. Epidermal peroxisome proliferator-activated receptor gamma as a target for ultraviolet B radiation. J. Biol. Chem. 2005, 280, 73–79. [Google Scholar]
- Konger, R.L.; Martel, K.C.; Jernigan, D.; Zhang, Q.; Travers, J.B. The peroxisome proliferator-activated receptor gamma system regulates ultraviolet B-induced prostaglandin e(2) production in human epidermal keratinocytes. PPAR Res. 2010, 2010, e467053. [Google Scholar]
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808. [Google Scholar] [CrossRef]
- Aouadi, M.; Laurent, K.; Prot, M.; le Marchand-Brustel, Y.; Binétruy, B.; Bost, F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes 2006, 55, 281–289. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Chen, C.; Li, X.; Gao, Y.; Li, P.; Zhang, Y.; Long, M.; Wang, Z.; Liu, G. Effect of insulin-like growth factor-1 (IGF-1) on the gluconeogenesis in calf hepatocytes cultured in vitro. Mol. Cell. Biochem. 2012, 362, 87–91. [Google Scholar] [CrossRef]
- Li, H.; Jogl, G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). J. Biol. Chem. 2009, 284, 1748–1754. [Google Scholar]
- Berkers, C.R.; Maddocks, O.D.K.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef]
- Levine, A.J.; Feng, Z.; Mak, T.W.; You, H.; Jin, S. Coordination and communication between the p53 and IGF-1–AKT–TOR signal transduction pathways. Genes Dev. 2006, 20, 267–275. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef]
- Goh, E.T.H.; Pardo, O.E.; Michael, N.; Niewiarowski, A.; Totty, N.; Volkova, D.; Tsaneva, I.R.; Seckl, M.J.; Gout, I. The involvement of hnRNP F in the regulation of cell proliferation via the mTOR/S6K2 pathway. J. Biol. Chem. 2010, 285, 17065–17076. [Google Scholar] [CrossRef]
- Harada, H.; Itasaka, S.; Kizaka-Kondoh, S.; Shibuya, K.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J. Biol. Chem. 2009, 284, 5332–5342. [Google Scholar]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Oxidative Phosphorylation. Available online: http://en.wikipedia.org/wiki/Oxidative_phosphorylation (accessed on 8 March 2013).
- Beta oxidation. Available online: http://en.wikipedia.org/wiki/Beta_oxidation (accessed on 8 March 2013).
- Searle, J.S.; Schollaert, K.L.; Wilkins, B.J.; Sanchez, Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat. Cell Biol. 2004, 6, 138–145. [Google Scholar] [CrossRef]
- Kotani, S.; Tugendreich, S.; Fujii, M.; Jorgensen, P.M.; Watanabe, N.; Hoog, C.; Hieter, P.; Todokoro, K. PKA and MPF-activated Polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1998, 1, 371–380. [Google Scholar] [CrossRef]
- Kishimoto, N.; Yamashita, I. Cyclic AMP regulates cell size of schizosaccharomyces pombe through CDC25 mitotic inducer. Yeast 2000, 16, 523–529. [Google Scholar] [CrossRef]
- Shukla, Y.; Reagan-Shaw, S.R.; Ahmad, N. Ultraviolet radiation causes induction of mitotic kinases Polo Like Kinase (Plk1) and aurora kinases-A and -B in HaCaT keratinocytes and SKH-1 hairless mouse skin: Relevance for skin carcinogenesis. Proc. Amer. Assoc. Cancer Res. 2005, 46, nr5693. [Google Scholar]
- Feng, Z. p53 Regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef]
- Ahmad, M.; Abdel-Wahab, Y.H.A.; Tate, R.; Flatt, P.R.; Pyne, N.J.; Furman, B.L. Effect of type-selective inhibitors on cyclic nucleotide phosphodiesterase activity and insulin secretion in the clonal insulin secreting cell line BRIN-BD11. Br. J. Pharmacol. 2000, 129, 1228–1234. [Google Scholar] [CrossRef]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef]
- Pichierri, P.; Rosselli, F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004, 23, 1178–1187. [Google Scholar] [CrossRef]
- Guo, F.; Li, J.; Du, W.; Zhang, S.; O’Connor, M.; Thomas, G.; Kozma, S.; Zingarelli, B.; Pang, Q.; Zheng, Y. mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells. Leukemia 2013, 27, 2040–2046. [Google Scholar] [CrossRef]
- Capoluongo, E. Insulin-like growth factor system and sporadic malignant melanoma. Am. J. Pathol. 2011, 178, 26–31. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Briviba, K.; Sies, H. Signaling by Singlet Oxygen in Biological Systems. In Antioxidant and Redox Regulation of Genes; Sen, C.K., Sies, H., Baeuerle, P.A., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 3–20. [Google Scholar]
- Tedesco, A.C.; Martínez, L.; González, S. Photochemistry and photobiology of actinic erythema: Defensive and reparative cutaneous mechanisms. Braz. J. Med. Biol. Res. 1997, 30, 561–575. [Google Scholar]
- Glickman, R.D. Phototoxicity to the retina: Mechanisms of damage. Int. J. Toxicol. 2002, 21, 473–490. [Google Scholar] [CrossRef]
- Glickman, R.D. Ultraviolet phototoxicity to the retina. Eye Contact Lens 2011, 37, 196–205. [Google Scholar] [CrossRef]
- Strozyk, E.; Kulms, D. The role of AKT/mTOR pathway in stress response to UV-Irradiation: Implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int. J. Mol. Sci. 2013, 14, 15260–15285. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A Complex interplay between Akt, TSC2, and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.-L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci.USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef]
- Tu, Y.; Yang, B.; Ji, C.; Yang, Z.; Gu, H.; Lu, C.-C.; Wang, R.; Su, Z.-L.; Chen, B.; Sun, W.-L.; et al. DNA-Dependent protein kinase catalytic subunit (DNA-PKcs)-SIN1 association mediates ultraviolet B (UVB)-induced Akt Ser-473 phosphorylation and skin cell survival. Mol. Cancer 2013, 12, e172. [Google Scholar] [CrossRef]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A double-edged sword in cell proliferation and genome stability. J. Oncol. 2012, 2012, e951724. [Google Scholar]
- Liu, S.; Bekker-Jensen, S.; Mailand, N.; Lukas, C.; Bartek, J.; Lukas, J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol. Cell. Biol. 2006, 26, 6056–6064. [Google Scholar] [CrossRef]
- Liu, K.; Graves, J.D.; Scott, J.D.; Li, R.; Lin, W.-C. Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol. Cell. Biol. 2013, 33, 4685–4700. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T. Phytochemicals as protectors against ultraviolet radiation: Versatility of effects and mechanisms. Planta Med. 2008, 74, 1548–1559. [Google Scholar] [CrossRef]
- Silveira, J.P.; Seito, L.N.; Eberlin, S.; Dieamant, G.C.; Nogueira, C.; Pereda, M.C.; Stasi, L.C.D. Photoprotective and antioxidant effects of rhubarb: Inhibitory action on tyrosinase and tyrosine kinase activities and TNF-Α, IL-1α and Α-MSH production in human melanocytes. BMC Complement. Altern. Med. 2013, 13, e49. [Google Scholar] [CrossRef]
- Foreback, J.L.; Sarma, V.; Yeager, N.R.; Younkin, E.M.; Remick, D.G.; Ward, P.A. Blood mononuclear cell production of TNF-alpha and IL-8: Engagement of different signal transduction pathways including the p42 MAP kinase pathway. J. Leukoc. Biol. 1998, 64, 124–133. [Google Scholar]
- Avalos-Díaz, E.; Alvarado-Flores, E.; Herrera-Esparza, R. UV-A irradiation induces transcriptionof IL-6 and TNF alpha genes in human keratinocytes and dermal fibroblasts. Rev. Rhum. Engl. Ed. 1999, 66, 13–19. [Google Scholar]
- Miller, C.C.; Hale, P.; Pentland, A.P. Ultraviolet B injury increases prostaglandin synthesis through a tyrosine kinase-dependent pathway. Evidence for UVB-induced epidermal growth factor receptor activation. J. Biol. Chem. 1994, 269, 3529–3533. [Google Scholar]
- Isoherranen, K.; Punnonen, K.; Jansen, C.; Uotila, P. Ultraviolet irradiation induces cyclooxygenase-2 expression in keratinocytes. Br. J. Dermatol. 1999, 140, 1017–1022. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar]
- Ramachandran, S.; Prasad, N.R.; Pugalendi, K.V.; Menon, V.P. Modulation of UVB-induced oxidative stress by ursolic acid in human blood lymphocytes. Asian J. Biochem. 2008, 3, 11–18. [Google Scholar]
- Zhu, Z.; Qian, Z.; Yan, Z.; Zhao, C.; Wang, H.; Ying, G. A Phase I pharmacokinetic study of ursolic acid nanoliposomes in healthy volunteers and patients with advanced solid tumors. Int. J. Nanomedicine 2013, 8, 129–136. [Google Scholar]
- Kunkel, S.D.; Elmore, C.J.; Bongers, K.S.; Ebert, S.M.; Fox, D.K.; Dyle, M.C.; Bullard, S.A.; Adams, C.M. Ursolic acid increases skeletal muscle and brown fat and decreases diet-induced obesity, glucose intolerance and fatty liver disease. PLoS One 2012, 7, e39332. [Google Scholar]
- Zhang, W.; Hong, D.; Zhou, Y.; Zhang, Y.; Shen, Q.; Li, J.; Hu, L.; Li, J. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim. Biophys. Acta 2006, 1760, 1505–1512. [Google Scholar]
- Arroba, A.I.; Revuelta-Cervantes, J.; Menes, L.; González-Rodríguez, Á.; Pardo, V.; de la Villa, P.; Burks, D.J.; Valverde, Á.M. Loss of protein tyrosine phosphatase 1B increases IGF-I receptor tyrosine phosphorylation but does not rescue retinal defects in IRS2-deficient mice. Invest. Ophthalmol. Vis. Sci. 2013, 54, 4215–4225. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; Nader, G.A. Ursolic acid directly promotes protein accretion in myotubes but does not affect myoblast proliferation. Cell Biochem. Funct. 2012, 30, 432–437. [Google Scholar] [CrossRef]
- Kunkel, S.D.; Suneja, M.; Ebert, S.M.; Bongers, K.S.; Fox, D.K.; Malmberg, S.E.; Alipour, F.; Shields, R.K.; Adams, C.M. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. 2011, 13, 627–638. [Google Scholar] [CrossRef]
- Rojo, A.I.; de Sagarra, M.R.; Cuadrado, A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.-Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One 2012, 7, e41831. [Google Scholar]
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D.D. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Signal. 2012, 17, 1670–1675. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, e200. [Google Scholar]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:Quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PLoS One 2012, 7, e44861. [Google Scholar] [CrossRef]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:Quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-Regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef]
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef]
- Hirota, A.; Kawachi, Y.; Itoh, K.; Nakamura, Y.; Xu, X.; Banno, T.; Takahashi, T.; Yamamoto, M.; Otsuka, F. Ultraviolet A irradiation induces NF-E2-related factor 2 activation in dermal fibroblasts: Protective role in UVA-induced apoptosis. J. Invest. Dermatol. 2005, 124, 825–832. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Hanneken, A.; Lin, F.-F.; Johnson, J.; Maher, P. Flavonoids protect human retinal pigment epithelial cells from oxidative-stress-induced death. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3164–3177. [Google Scholar] [CrossRef]
- Hartley, D.; Cooper, G.M. Role of mTOR in the degradation of IRS-1: Regulation of PP2A activity. J. Cell. Biochem. 2002, 85, 304–314. [Google Scholar] [CrossRef]
- King, E.R.; Wong, K.-K. Insulin-like growth factor: Current concepts and new developments in cancer therapy. Recent Pat. Anticancer Drug Discov. 2012, 7, 14–30. [Google Scholar] [CrossRef]
- Gottlieb, E.; Vousden, K.H. p53 Regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2010, 2, a001040. [Google Scholar]
- Li, Y.; Inoki, K.; Vacratsis, P.; Guan, K.-L. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14–3-3. J. Biol. Chem. 2003, 278, 13663–13671. [Google Scholar]
- Hernández, G.; Lal, H.; Fidalgo, M.; Guerrero, A.; Zalvide, J.; Force, T.; Pombo, C.M. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 2011, 317, 2938–2949. [Google Scholar] [CrossRef]
- Johnson, R.F.; Witzel, I.-I.; Perkins, N.D. p53-Dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef]
- Choi, S.M.; Tucker, D.F.; Gross, D.N.; Easton, R.M.; DiPilato, L.M.; Dean, A.S.; Monks, B.R.; Birnbaum, M.J. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. CellBiol. 2010, 30, 5009–5020. [Google Scholar]
- Wan, Z.; Root-McCaig, J.; Castellani, L.; Kemp, B.E.; Steinberg, G.R.; Wright, D.C. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 2014, 22, 730–738. [Google Scholar]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Jung, S.H.; Ha, Y.J.; Shim, E.K.; Choi, S.Y.; Jin, J.L.; Yun-Choi, H.S.; Lee, J.R. Insulin-mimetic and insulin-sensitizing activities of a pentacyclic triterpenoid insulin receptor activator. Biochem. J. 2007, 403, 243–250. [Google Scholar] [CrossRef]
- Pagel-Langenickel, I.; Bao, J.; Joseph, J.J.; Schwartz, D.R.; Mantell, B.S.; Xu, X.; Raghavachari, N.; Sack, M.N. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 2008, 283, 22464–22472. [Google Scholar] [CrossRef]
- Medina-Gomez, G.; Gray, S.; Vidal-Puig, A. Adipogenesis and lipotoxicity: Role of peroxisome proliferator-activated receptor gamma (PPARgamma) and PPARgamma coactivator-1 (PGC1). Public Health Nutr. 2007, 10, 1132–1137. [Google Scholar]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar]
- Im, S.-S.; Kim, M.-Y.; Kwon, S.-K.; Kim, T.-H.; Bae, J.-S.; Kim, H.; Kim, K.-S.; Oh, G.-T.; Ahn, Y.-H. Peroxisome proliferator-activated receptor is responsible for the up-regulation of hepatic glucose-6-phosphatase gene expression in fasting and Db/db mice. J. Biol. Chem. 2010, 286, 1157–1164. [Google Scholar]
- Summermatter, S.; Santos, G.; Pérez-Schindler, J.; Handschin, C. Skeletal muscle PGC-1α controls whole-body lactate homeostasis through estrogen-related receptor α-dependent activation of LDH B and repression of LDH A. Proc. Natl. Acad. Sci.USA 2013, 110, 8738–8743. [Google Scholar]
- Wende, A.R.; Schaeffer, P.J.; Parker, G.J.; Zechner, C.; Han, D.-H.; Chen, M.M.; Hancock, C.R.; Lehman, J.J.; Huss, J.M.; McClain, D.A.; et al. A Role for the transcriptional coactivator PGC-1alpha in muscle refueling. J. Biol. Chem. 2007, 282, 36642–36651. [Google Scholar]
- Kim, H.-J.; Jee, H.J.; Yun, J. DNA damage induces down-regulation of PEPCK and G6P gene expression through degradation of PGC-1alpha. Acta Biochim. Biophys. Sin. 2011, 43, 589–594. [Google Scholar]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef]
- Pohnke, Y.; Schneider-Merck, T.; Fahnenstich, J.; Kempf, R.; Christian, M.; Milde-Langosch, K.; Brosens, J.J.; Gellersen, B. Wild-type p53 protein is up-regulated upon cyclic adenosine monophosphate-induced differentiation of human endometrial stromal cells. J. Clin. Endocrinol. Metab. 2004, 89, 5233–5244. [Google Scholar] [CrossRef]
- Destefano, M.A.; Jacinto, E. Regulation of insulin receptor substrate-1 by mTORC2 (mammalian target of rapamycin complex 2). Biochem. Soc. Trans. 2013, 41, 896–901. [Google Scholar] [CrossRef]
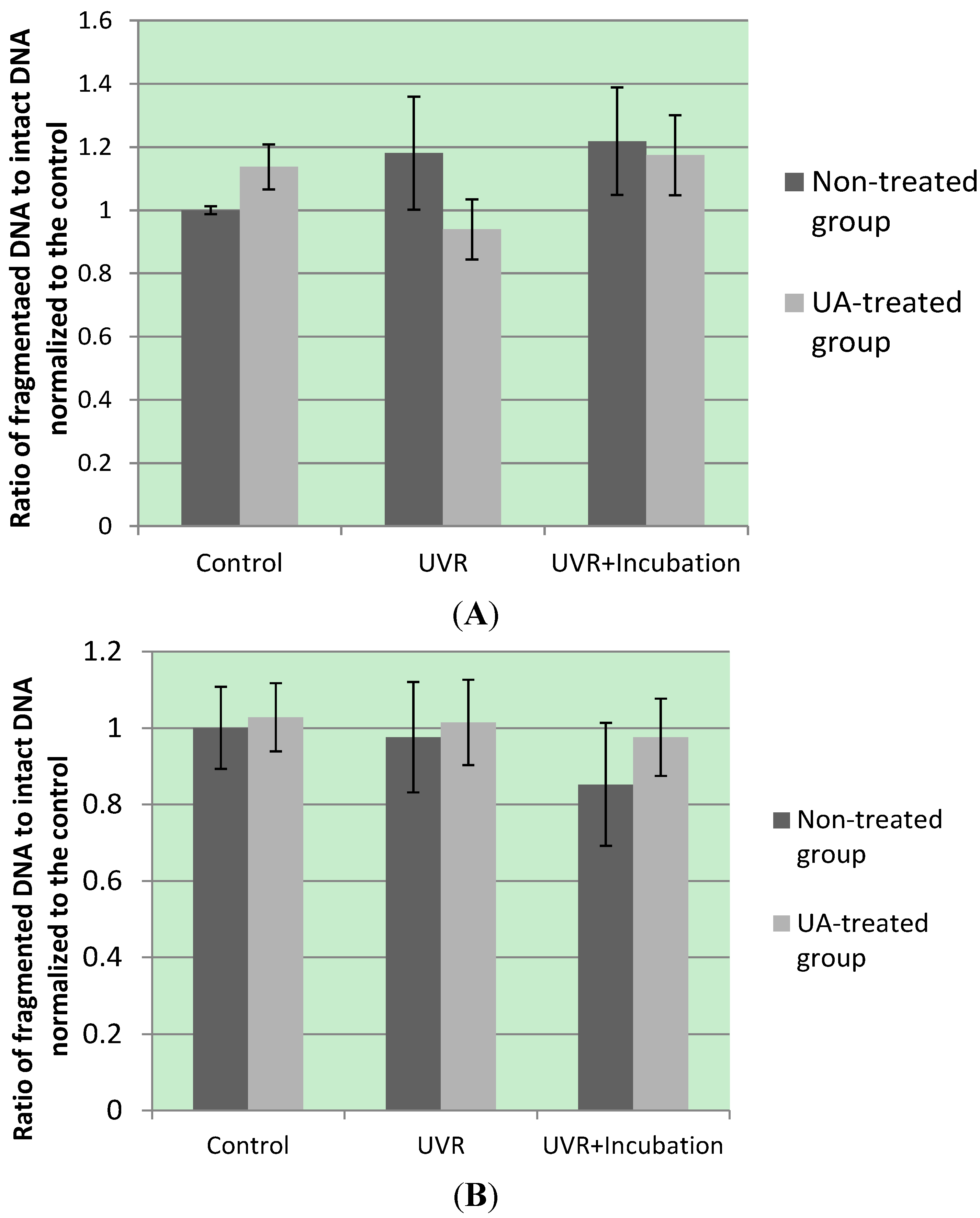
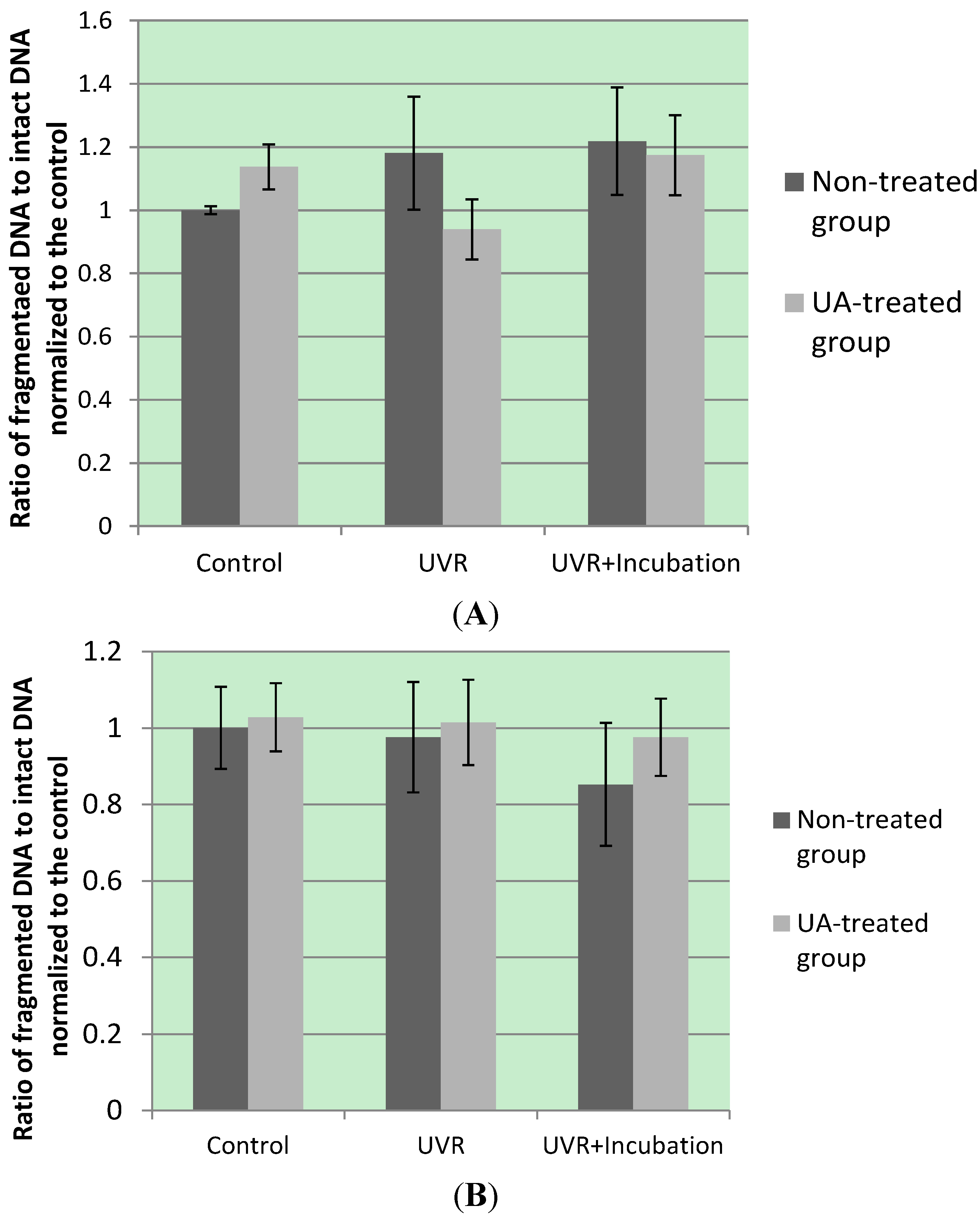
- Qualitative analysis of DNA fragmentation by agarose gel electrophoresis. Dompé Research Center: L'Aquila, Italy. Available online: http://www.immunologia.unimore.it/SITO%20AC/APOBOOK98/CHAP4.HTM (accessed on 11 May 2011).
- Lou, L.; Urbani, J.; Ribeiro-Neto, F.; Altschuler, D.L. cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. J. Biol. Chem. 2002, 277, 32799–32806. [Google Scholar]
- Valle, I.; Álvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Li, Y.; Kang, Z.; Li, S.; Kong, T.; Liu, X.; Sun, C. Ursolic Acid stimulates lipolysis in primary-cultured Rat adipocytes. Mol. Nutr. Food Res. 2010, 54, 1609–1617. [Google Scholar] [CrossRef]
- Naderi, E.H.; Findley, H.W.; Ruud, E.; Blomhoff, H.K.; Naderi, S. Activation of cAMP signaling inhibits DNA damage-induced apoptosis in BCP-ALL Cells through abrogation of p53 accumulation. Blood 2009, 114, 608–618. [Google Scholar] [CrossRef]
- Honnor, R.C.; Dhillon, G.S.; Londos, C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. J. Biol. Chem. 1985, 260, 15122–15129. [Google Scholar]
- National Toxicology Program. Report on Carcinogens, 12th ed.Public Health Service, U.S. Department of Health and Human Services, 2011; pp. 429–430. Available online: http://ntp.niehs.nih.gov/ntp/roc/twelfth/profiles/UltravioletRadiationRelatedExposures.pdf (accessed on 2 December 2012).
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.-L.; Ordóñez, G.R.; Bignell, G.R.; et al. A Comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Rodríguez-Escudero, I.; Oliver, M.D.; Andrés-Pons, A.; Molina, M.; Cid, V.J.; Pulido, R. A comprehensive functional analysis of PTEN mutations: Implications in tumor- and autism-related syndromes. Hum. Mol. Genet. 2011, 20, 4132–4142. [Google Scholar] [CrossRef]
- Gagliardi, P.A.; di Blasio, L.; Orso, F.; Seano, G.; Sessa, R.; Taverna, D.; Bussolino, F.; Primo, L. 3-Phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia N. Y. 2012, 14, 719–731. [Google Scholar]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Chu, W.; Guan, K.; Wan, Y. AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes. J. Biol. Chem. 2008, 283, 28897–28908. [Google Scholar] [CrossRef]
- Yao, J.; Bi, H.-E.; Sheng, Y.; Cheng, L.-B.; Wendu, R.-L.; Wang, C.-H.; Cao, G.-F.; Jiang, Q. Ultraviolet (UV) and hydrogen peroxide activate ceramide-ER stress-AMPK signaling axis to promote retinal pigment epithelium (RPE) cell apoptosis. Int. J. Mol. Sci. 2013, 14, 10355–10368. [Google Scholar]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Eisele, P.S.; Salatino, S.; Sobek, J.; Hottiger, M.O.; Handschin, C. The PGC-1 coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 2013, 288, 2246–2260. [Google Scholar]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, Y.-H.; Sun, Y.; Glickman, R.D. Ursolic Acid-Regulated Energy Metabolism—Reliever or Propeller of Ultraviolet-Induced Oxidative Stress and DNA Damage? Proteomes 2014, 2, 399-425. https://doi.org/10.3390/proteomes2030399
Lee Y-H, Sun Y, Glickman RD. Ursolic Acid-Regulated Energy Metabolism—Reliever or Propeller of Ultraviolet-Induced Oxidative Stress and DNA Damage? Proteomes. 2014; 2(3):399-425. https://doi.org/10.3390/proteomes2030399
Chicago/Turabian StyleLee, Yuan-Hao, Youping Sun, and Randolph D. Glickman. 2014. "Ursolic Acid-Regulated Energy Metabolism—Reliever or Propeller of Ultraviolet-Induced Oxidative Stress and DNA Damage?" Proteomes 2, no. 3: 399-425. https://doi.org/10.3390/proteomes2030399
APA StyleLee, Y.-H., Sun, Y., & Glickman, R. D. (2014). Ursolic Acid-Regulated Energy Metabolism—Reliever or Propeller of Ultraviolet-Induced Oxidative Stress and DNA Damage? Proteomes, 2(3), 399-425. https://doi.org/10.3390/proteomes2030399