
Azidohomoalanine (AHA) Metabolic Labeling Reveals Unique Proteomic Insights into Protein Synthesis and Degradation in Response to Bortezomib Treatment


Abstract
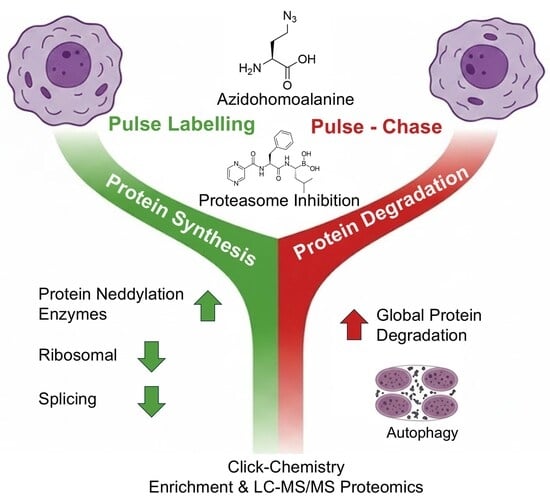
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals
2.3. Cell Viability Assays
2.4. Azidohomoalanine (AHA) Pulse–Chase Method
2.5. Biotinylated Protein Capture
2.6. Mass Spectrometry for AHA Labeling
2.7. Label-Free LC-MS/MS
2.8. Statistics and Data Analysis
2.9. Western Blot Analysis
3. Results
3.1. Characterization of Bortezomib Treatment on Cell Viability
3.2. Nascent Peptide Proteomics
3.3. Proteins Exhibiting Increased Synthesis upon Bortezomib Treatment
3.4. Proteins Exhibiting Decreased Synthesis upon Bortezomib Treatment
3.5. Pulse–Chase Protein Degradation Proteomics
3.6. Determination of Protein Half-Life After AHA Pulse–Chase
3.7. Diverse Protein Groups Degraded upon Bortezomib Treatment
3.8. Investigating the Role of Autophagy in Protein Degradation
3.9. Label-Free Proteomic Analysis
4. Discussion
4.1. Impact of Nascent Protein Synthesis
4.2. Increased Synthesis of Proteins
Protein Neddylation
4.3. Decreased Protein Synthesis upon Bortezomib Treatment
4.4. Protein Degradation
4.5. Stabilized Proteins upon Bortezomib Treatment
4.6. Destabilized Proteins upon Bortezomib Treatment
4.7. Investigating Autophagy and Proteasome Inhibition
4.8. Functional Outcomes of Drug Treatments
4.9. Future Considerations and Caveats
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ho, M.; Bianchi, G.; Anderson, K.C. Proteomics-inspired precision medicine for treating and understanding multiple myeloma. Expert Rev. Precis. Med. Drug Dev. 2020, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Cumova, J.; Potacova, A.; Zdrahal, Z.; Hajek, R. Proteomic Analysis in Multiple Myeloma Research. Mol. Biotechnol. 2011, 47, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Shahshahan, M.A.; Beckley, M.N.; Jazirehi, A.R. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: Basic and clinical aspects. Am. J. Cancer Res. 2011, 1, 913–924. [Google Scholar] [PubMed]
- Curran, M.P.; McKeage, K. Bortezomib. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef]
- Tyrna, P.; Procyk, G.; Szeleszczuk, Ł.; Mlynarczuk-Bialy, I. Different Strategies to Overcome Resistance to Proteasome Inhibitors—A Summary 20 Years after Their Introduction. Int. J. Mol. Sci. 2024, 25, 8949. [Google Scholar] [CrossRef]
- Cvek, B.; Dvorak, Z. The ubiquitin-proteasome system (UPS) and the mechanism of action of bortezomib. Curr. Pharm. Des. 2011, 17, 1483–1499. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, C.; Lu, D.; Wu, X.; Gao, L.; Zhang, W. Endoplasmic reticulum stress and autophagy participate in apoptosis induced by bortezomib in cervical cancer cells. Biotechnol. Lett. 2016, 38, 357–365. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Gasparri, F.; Galvani, A.; Nici, L.; Darnowski, J.W.; Barbone, D.; Fennell, D.A.; Gaudino, G.; Porta, C.; Mutti, L. Bortezomib inhibits nuclear factor-κB dependent survival and has potent in vivo activity in mesothelioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5942–5951. [Google Scholar] [CrossRef]
- Rastogi, N.; Mishra, D.P. Therapeutic targeting of cancer cell cycle using proteasome inhibitors. Cell Div. 2012, 7, 26. [Google Scholar] [CrossRef]
- Kubicki, T.; Bednarek, K.; Kostrzewska-Poczekaj, M.; Luczk, M.; Lewandowski, K.; Gil, L.; Jarmuz-Szymczak, M.; Dytfeld, D. Bortezomib- and carfilzomib-resistant myeloma cells show increased activity of all three arms of the unfolded protein response. Am. J. Cancer Res. 2022, 12, 3280–3293. [Google Scholar]
- Tierney, C.; Bazou, D.; Majumder, M.M.; Anttila, P.; Silvennoinen, R.; Heckman, C.A.; Dowling, P.; O’Gorman, P. Next generation proteomics with drug sensitivity screening identifies sub-clones informing therapeutic and drug development strategies for multiple myeloma patients. Sci. Rep. 2021, 11, 12866. [Google Scholar] [CrossRef]
- Ma, Y.; McClatchy, D.B.; Barkallah, S.; Wood, W.W.; Yates, J.R., III. Quantitative analysis of newly synthesized proteins. Nat. Protoc. 2018, 13, 1744–1762. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.A.; Eldeeb, M.A.; Wuest, M.; Mercer, J.; Fahlman, R.P. Proteomic characterization of EL4 lymphoma-derived tumors upon chemotherapy treatment reveals potential roles for lysosomes and caspase-6 during tumor cell death in vivo. Proteomics 2017, 17, 1700060. [Google Scholar] [CrossRef] [PubMed]
- Pascovici, D.; Handler, D.C.L.; Wu, J.X.; Haynes, P.A. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 2016, 16, 2448–2453. [Google Scholar] [CrossRef]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Lai, X.; Huang, C.; Nie, X.; Chen, Q.; Tang, Y.; Fu, X.; Lin, Y.; Nie, C.; Xu, X.; Wang, X.; et al. Bortezomib Inhibits Multiple Myeloma Cells by Transactivating ATF3 to Trigger miR-135a-5p-Dependent Apoptosis. Front. Oncol. 2021, 11, 720261. [Google Scholar] [CrossRef]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef]
- Berg Luecke, L.; Gundry, R.L. Assessment of Streptavidin Bead Binding Capacity to Improve Quality of Streptavidin-based Enrichment Studies. J. Proteome Res. 2021, 20, 1153–1164. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Johnson, D.E. Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via JNK activation. Cancer Lett. 2012, 314, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.M.P.; de Sousa, R.W.R.; de Oliveira Ferreira, J.R.; Militao, G.C.G.; Bezerra, D.P. Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharmacol. Res. 2021, 168, 105582. [Google Scholar] [CrossRef]
- Dai, Y.; Guo, X.; Yang, C. Effect of bortezomib on proliferation and apoptosis of myeloma cells by activating Wnt/β-catenin signaling pathway. Oncol. Lett. 2020, 20, 1295–1299. [Google Scholar] [CrossRef]
- Xia, Y.; Yan, L.H.; Huang, B.; Liu, M.; Liu, X.; Huang, C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J. Neurochem. 2014, 129, 99–106. [Google Scholar] [CrossRef]
- Seibold, M.; Stühmer, T.; Kremer, N.; Mottok, A.; Scholz, C.-J.; Schlosser, A.; Leich, E.; Holzgrabe, U.; Brunnert, D.; Barrio, S.; et al. RAL GTPases mediate multiple myeloma cell survival and are activated independently of oncogenic RAS. Haematologica 2020, 105, 2316–2326. [Google Scholar] [CrossRef]
- Huang, X.-D.; Du, L.; Cheng, X.-C.; Lu, Y.-X.; Liu, Q.-W.; Wang, Y.-W.; Liao, Y.-J.; Lin, D.-D.; Xiao, F.-J. OTUB1/NDUFS2 axis promotes pancreatic tumorigenesis through protecting against mitochondrial cell death. Cell Death Discov. 2024, 10, 190. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Q.; Li, T.; Qian, J.; Lu, Y.; Li, Y.; Bi, E.; Reu, F.; Qin, Y.; Drazba, J.; et al. Role of Myeloma-Derived MIF in Myeloma Cell Adhesion to Bone Marrow and Chemotherapy Response. J. Natl. Cancer Inst. 2016, 108, djw131. [Google Scholar] [CrossRef]
- Gu, Y.; Kaufman, J.L.; Bernal, L.; Torre, C.; Matulis, S.M.; Harvey, R.D.; Chen, J.; Sun, S.-Y.; Boise, L.H.; Lonial, S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 2014, 123, 3269–3276. [Google Scholar] [CrossRef]
- Maghames, C.M.; Lobato-Gil, S.; Perrin, A.; Trauchessec, H.; Rodriguez, M.S.; Urbach, S.; Marin, P.; Xirodimas, D.P. NEDDylation promotes nuclear protein aggregation and protects the Ubiquitin Proteasome System upon proteotoxic stress. Nat. Commun. 2018, 9, 4376. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Jacobs, H.M.; Delmore, J.E.; Buon, L.; Hunter, Z.R.; Monrose, V.; Yu, J.; Smith, P.G.; Richardson, P.G.; Anderson, K.C.; et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther. 2012, 11, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Largo, C.; Alvarez, S.; Saez, B.; Blesa, D.; Martin-Subero, J.I.; Gonzalez-Garcia, I.; Brieva, J.A.; Dopazo, J.; Siebert, R.; Calasanz, M.J.; et al. Identification of overexpressed genes in frequently gained/amplified chromosome regions in multiple myeloma. Haematologica 2006, 91, 184–191. [Google Scholar] [PubMed]
- Chen, Y.; Peng, C.; Tan, W.; Yu, J.; Zayas, J.; Peng, Y.; Lou, Z.; Pei, H.; Wang, L. Tumor protein D52 (TPD52) affects cancer cell metabolism by negatively regulating AMPK. Cancer Med. 2022, 12, 488–499. [Google Scholar] [CrossRef]
- Tiacci, E.; Orvietani, P.-L.; Bigerna, B.; Pucciarini, A.; Corthals, G.L.; Pettirossi, V.; Martelli, M.P.; Liso, A.; Benedetti, R.; Pacini, R.; et al. Tumor protein D52 (TPD52): A novel B-cell/plasma-cell molecule with unique expression pattern and Ca2+-dependent association with annexin VI. Blood 2005, 105, 2812–2820. [Google Scholar] [CrossRef]
- Chatterjee, M.; Rancso, C.; Stühmer, T.; Eckstein, N.; Andrulis, M.; Gerecke, C.; Lorentz, H.; Royer, H.-D.; Bargou, R.C. The Y-box binding protein YB-1 is associated with progressive disease and mediates survival and drug resistance in multiple myeloma. Blood 2008, 111, 3714–3722. [Google Scholar] [CrossRef]
- Bommert, K.; Effenberger, M.; Leich, E.; Kuspert, M.; Murphy, D.; Langer, C.; Moll, R.; Janz, S.; Mottok, A.; Weissbach, S.; et al. The feed-forward loop between YB-1 and MYC is essential for multiple myeloma cell survival. Leukemia 2013, 27, 441–450. [Google Scholar] [CrossRef]
- Gahlot, P.; Kravic, B.; Rota, G.; van den Boom, J.; Levantovsky, S.; Schulze, N.; Maspero, E.; Polo, S.; Behrends, C.; Meyer, H. Lysosomal damage sensing and lysophagy initiation by SPG20-ITCH. Mol. Cell 2024, 84, 1556–1569.e10. [Google Scholar] [CrossRef]
- Li, N.; Deng, L.; Zhang, Y.; Tang, X.; Lei, B.; Zhang, Q. IGF2BP2 modulates autophagy and serves as a prognostic marker in glioma. iBrain 2024, 10, 19–33. [Google Scholar] [CrossRef]
- Neuffer, S.J.; Beltran-Cardona, D.; Jimenez-Perez, K.; Clancey, L.; Brown, A.; New, L.; Cooper, C.D. AP-3 complex subunit delta gene, ap3d1, regulates melanogenesis and melanophore survival via autophagy in zebrafish (Danio rerio). Pigment Cell Melanoma Res. 2022, 35, 495–505. [Google Scholar] [CrossRef]
- Li, K.; Yang, L.; Zhang, C.; Niu, Y.; Li, W.; Liu, J.-J. HPS6 interacts with dynactin p150Glued to mediate retrograde trafficking and maturation of lysosomes. J. Cell Sci. 2014, 127, 4574–4588. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Wang, L.; Chou, C.-H.; Liu, K.; Nakata, T.; Jaiswal, A.; Yao, J.; Lefkovith, A.; Omar, A.; Perrigoue, J.G.; et al. QRICH1 Dictates the Outcome of ER Stress through Transcriptional Control of Proteostasis. Science 2021, 371, eabb6896. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, P.W.; Geiger, J.D.; Chen, X. Overcoming Chemoresistance: Altering pH of Cellular Compartments by Chloroquine and Hydroxychloroquine. Front. Cell Dev. Biol. 2021, 9, 627639. [Google Scholar] [CrossRef]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjorkoy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef]
- Hendrix, R.A.; Saunders, W.H. Cervical meningioma: Report of a case and review of the literature. Trans.-Pa. Acad. Ophthalmol. Otolaryngol. 1985, 37, 235–239. [Google Scholar]
- Cross, J.; Durgan, J.; McEwan, D.G.; Tayler, M.; Ryan, K.M.; Florey, O. Lysosome damage triggers direct ATG8 conjugation and ATG2 engagement via non-canonical autophagy. J. Cell Biol. 2023, 222, e202303078. [Google Scholar] [CrossRef]
- Zhang, R.; Torraca, V.; Lyu, H.; Xiao, S.; Guo, D.; Zhou, C.; Tang, J. RUNDC1 negatively mediates the fusion of autophagosomes with lysosomes via regulating SNARE complex assembly. Autophagy 2024, 20, 454–456. [Google Scholar] [CrossRef]
- Kankanamalage, S.G.; Lee, A.-Y.; Wichaidit, C.; Lorente-Rodriguez, A.; Shah, A.M.; Stippec, S.; Whitehurst, A.W.; Cobb, M.H. Multistep regulation of autophagy by WNK1. Proc. Natl. Acad. Sci. USA 2016, 113, 14342–14347. [Google Scholar] [CrossRef]
- Kankanamalage, S.G.; Lee, A.-Y.; Wichaidit, C.; Lorente-Rodriguez, A.; Shah, A.M.; Stippec, S.; Whitehurst, A.W.; Cobb, M.H. WNK1 is an unexpected autophagy inhibitor. Autophagy 2017, 13, 969–970. [Google Scholar] [CrossRef]
- Ye, Y.; Tyndall, E.R.; Bui, V.; Tang, Z.; Shen, Y.; Jiang, X.; Flanagan, J.M.; Wang, H.-G.; Tian, F. An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy. Nat. Commun. 2021, 12, 374. [Google Scholar] [CrossRef] [PubMed]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, Y.; He, C.; Zhang, X.; Torraca, V.; Wang, S.; Liu, N.; Yang, J.; Liu, S.; Yuan, J.; et al. RUNDC1 inhibits autolysosome formation and survival of zebrafish via clasping ATG14-STX17-SNAP29 complex. Cell Death Differ. 2023, 30, 2231–2248. [Google Scholar] [CrossRef] [PubMed]
- Goebel, T.; Mausbach, S.; Tuermer, A.; Eltahir, H.; Winter, D.; Gieselmann, V.; Thelen, M. Proteaphagy in Mammalian Cells Can Function Independent of ATG5/ATG7. Mol. Cell. Proteom. 2020, 19, 1120–1131. [Google Scholar] [CrossRef]
- Anim, E.P.; Mezzanotte, J.; Chu, S.; Stochaj, U. Bioorthogonal Non-Canonical Amino Acid Tagging (BONCAT) to detect newly synthesized proteins in cells and their secretome. PLoS ONE 2025, 20, e0329857. [Google Scholar] [CrossRef]
- Hooshmandi, M.; Sharma, V.; Perez, C.T.; Sood, R.; Krimbacher, K.; Wong, C.; Lister, K.C.; Guzman, A.U.; Bartley, T.D.; Rocha, C.; et al. Excitatory neuron-specific suppression of the integrated stress response contributes to autism-related phenotypes in fragile X syndrome. Neuron 2023, 111, 3028–3040. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Tian, Y.; Yang, J.; Jia, Y.; Xie, Y.; Cheng, L.; Chen, S.; Wu, L.; Qin, Y.; et al. Srsf3-Dependent APA Drives Macrophage Maturation and Limits Atherosclerosis. Circ. Res. 2025, 126, 985–1009. [Google Scholar] [CrossRef]
- Jecmen, T.; Tuzhilkin, R.; Sulc, M. Photo-Methionine Azidohomoalanine Homopropargylglycine are Incorporated into Newly Synthesized Proteins at Different Rates Differentially Affect the Growth Protein Expression Levels of Auxotrophic Prototrophic, E. coli in Minimal Medium. Int. J. Mol. Sci. 2023, 24, 11779. [Google Scholar] [CrossRef]
- Kirscher, F.; Arnold-Schilid, D.; Leps, C.; Lacki, M.K.; Klein, M.; Chen, Y.; Ludt, A.; Marini, F.; Kucuk, C.; Stein, L.; et al. Modulation of cellular transcriptome and proteome composition by azidohomoalanine-implications on click chemistry-based secretome analysis. J. Mol. Med. 2023, 101, 855–867. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Protein | Description | DMSO ½-Life (h) | Bortezomib ½-Life (h) |
|---|---|---|---|---|
| Q7Z5L9 | IRF2BP2 | Interferon regulatory factor 2-binding protein | 2.5 | 6.1 |
| P30049 | ATP5F1D | ATP synthase subunit delta | 1.1 | 3.6 |
| Q2TAL8 | QRICH1 | Transcriptional regulator QRICH1 | 2.2 | 7.6 |
| P53990 | IST1 | IST1 homolog | 6.8 | 25.6 |
| O14617 | AP3D1 | AP-3 complex subunit delta-1 | 1.9 | 7.7 |
| Q9Y2Z4 | YARS2 | Tyrosine–tRNA ligase, mitochondrial | 5.2 | 25.6 |
| Q86YV9 | HPS6 | BLOC-2 complex member HPS6 | 4.1 | 25.6 |
| Q9H4A5 | GOLPH3L | Golgi phosphoprotein 3-like | 2.7 | 25.6 |
| Q8IY17 | PNPLA6 | Patatin-like phospholipase domain-containing protein 6 | 1.5 | 25.6 |
| P18669 | PGAM1 | Phosphoglycerate mutase 1 | 1.4 | 25.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhourani, L.; Tabana, Y.; Anand, A.; Fahlman, R.P. Azidohomoalanine (AHA) Metabolic Labeling Reveals Unique Proteomic Insights into Protein Synthesis and Degradation in Response to Bortezomib Treatment. Proteomes 2025, 13, 63. https://doi.org/10.3390/proteomes13040063
Alhourani L, Tabana Y, Anand A, Fahlman RP. Azidohomoalanine (AHA) Metabolic Labeling Reveals Unique Proteomic Insights into Protein Synthesis and Degradation in Response to Bortezomib Treatment. Proteomes. 2025; 13(4):63. https://doi.org/10.3390/proteomes13040063
Chicago/Turabian StyleAlhourani, Lina, Yasser Tabana, Ashwin Anand, and Richard P. Fahlman. 2025. "Azidohomoalanine (AHA) Metabolic Labeling Reveals Unique Proteomic Insights into Protein Synthesis and Degradation in Response to Bortezomib Treatment" Proteomes 13, no. 4: 63. https://doi.org/10.3390/proteomes13040063
APA StyleAlhourani, L., Tabana, Y., Anand, A., & Fahlman, R. P. (2025). Azidohomoalanine (AHA) Metabolic Labeling Reveals Unique Proteomic Insights into Protein Synthesis and Degradation in Response to Bortezomib Treatment. Proteomes, 13(4), 63. https://doi.org/10.3390/proteomes13040063