

Comparative Proteome-Wide Abundance Profiling of Yeast Strains Deleted for Cdc48 Adaptors



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Yeast Strains, Growth Conditions and Protein Extraction
2.3. Protein Digestion, TMT Labeling and Sample Processing
2.4. Mass Spectrometry Data Acquisition and Processing
3. Results and Discussion
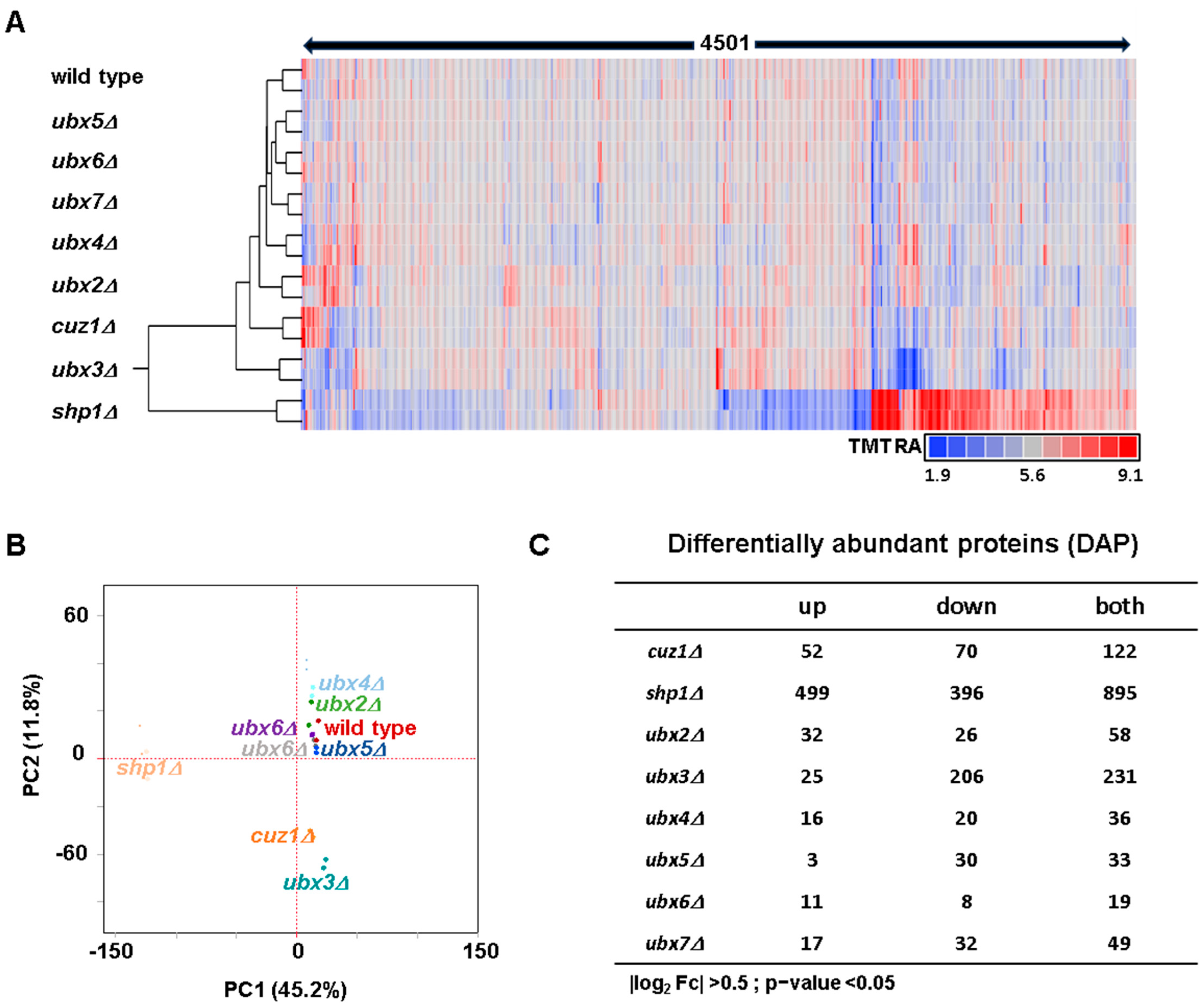
3.1. Analysis of the Differences in Protein Abundance Between the Strains Lacking Cdc48 Adaptors and the Wild Type Strain Revealed Specific Proteome Level Abundance Changes
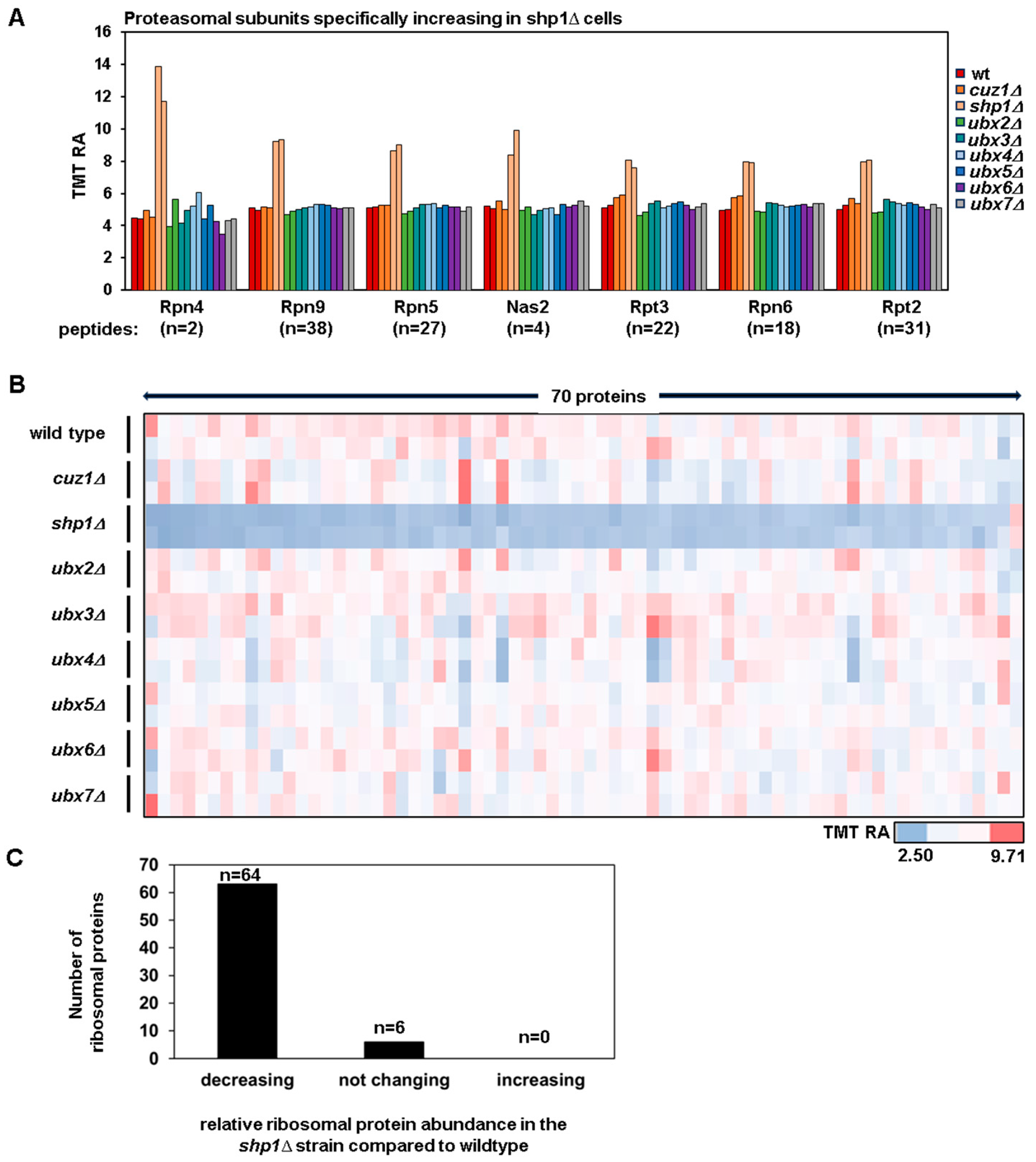
3.2. The Upregulation of the Transcription Factor Rpn4 and the Downregulation of Ribosomal Proteins Suggest the Presence of Proteotoxic Stress in the shp1Δ Strain
3.3. A Specific Subset of Mitochondrial Proteins Is Downregulated in the ubx3Δ and cuz1Δ Strains
3.4. Examples of Proteins That Change Specifically in Cdc48 Adaptor Protein Deletion Strains
4. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. Regulation of Signaling by Non-degradative Ubiquitination. J. Biol. Chem. 2009, 284, 8209. [Google Scholar] [CrossRef]
- Liao, Y.; Sumara, I.; Pangou, E. Non-proteolytic ubiquitylation in cellular signaling and human disease. Commun. Biol. 2022, 5, 114. [Google Scholar] [CrossRef]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef]
- Hirabayashi, M.; Inoue, K.; Tanaka, K.; Nakadate, K.; Ohsawa, Y.; Kamei, Y.; Popiel, A.H.; Sinohara, A.; Iwamatsu, A.; Kimura, Y.; et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001, 8, 977–984. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Columbres, R.C.A.; Chin, Y.; Pratti, S.; Quinn, C.; Gonzalez-Cuyar, L.F.; Weiss, M.; Quintero-Rivera, F.; Kimonis, V. Novel Variants in the VCP Gene Causing Multisystem Proteinopathy 1. Genes 2023, 14, 676. [Google Scholar] [CrossRef] [PubMed]
- Iannibelli, E.; Gibertini, S.; Cheli, M.; Blasevich, F.; Cavaliere, A.; Riolo, G.; Ruggieri, A.; Maggi, L. VCP-related myopathy: A case series and a review of literature. Acta Myol. 2023, 42, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Capone, F.; Polo, A.; Bagnara, P.; Budillon, A. Valosin-Containing Protein (VCP)/p97: A Prognostic Biomarker and Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 10177. [Google Scholar] [CrossRef] [PubMed]
- Kilgas, S.; Ramadan, K. Inhibitors of the ATPase p97/VCP: From basic research to clinical applications. Cell Chem. Biol. 2023, 30, 3–21. [Google Scholar] [CrossRef]
- Vekaria, P.H.; Home, T.; Weir, S.; Schoenen, F.J.; Rao, R. Targeting p97 to Disrupt Protein Homeostasis in Cancer. Front. Oncol. 2016, 6, 181. [Google Scholar] [CrossRef]
- Benajiba, L.; Carraway, H.E.; Hamad, N.; Stein, E.M.; Yannakou, C.K.; Burroughs, A.; Harris, S.; Lane, H.; Nguyen, D.D.; Stuart, M.; et al. Trials in Progress: A Phase I Study to Evaluate the Safety and Pharmacokinetic Profiles of CB-5339 in Participants with Relapsed/Refractory Acute Myeloid Leukemia or Relapsed/Refractory Intermediate or High-Risk Myelodysplastic Syndrome. Blood 2020, 136, 21. [Google Scholar] [CrossRef]
- Stolz, A.; Hilt, W.; Buchberger, A.; Wolf, D.H. Cdc48: A power machine in protein degradation. Trends Biochem. Sci. 2011, 36, 515–523. [Google Scholar] [CrossRef]
- Ye, Y. Diverse functions with a common regulator: Ubiquitin takes command of an AAA ATPase. J. Struct. Biol. 2006, 156, 29–40. [Google Scholar] [CrossRef]
- Shcherbik, N.; Haines, D.S. Cdc48p(Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol. Cell 2007, 25, 385–397. [Google Scholar] [CrossRef]
- Olszewski, M.M.; Williams, C.; Dong, K.C.; Martin, A. The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol. 2019, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Anton, V. Mitochondrial Surveillance by Cdc48/p97: MAD vs. Membrane Fusion. Int. J. Mol. Sci. 2020, 21, 6841. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-L.; Chen, R.-H. Assembly and quality control of the protein phosphatase 1 holoenzyme involves the Cdc48–Shp1 chaperone. J. Cell Sci. 2015, 128, 1180–1192. [Google Scholar] [CrossRef]
- Higgins, R.; Kabbaj, M.-H.; Sherwin, D.; Howell, L.A.; Hatcher, A.; Tomko, R.J.; Wang, Y. The Cdc48 Complex Alleviates the Cytotoxicity of Misfolded Proteins by Regulating Ubiquitin Homeostasis. Cell Rep. 2020, 32, 107898. [Google Scholar] [CrossRef]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef]
- Wolf, D.H.; Stolz, A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 117–124. [Google Scholar] [CrossRef]
- Cao, K.; Nakajima, R.; Meyer, H.H.; Zheng, Y. The AAA-ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell 2003, 115, 355–367. [Google Scholar] [CrossRef]
- Kloppsteck, P.; Ewens, C.A.; Förster, A.; Zhang, X.; Freemont, P.S. Regulation of p97 in the ubiquitin-proteasome system by the UBX protein-family. Biochim. Biophys. Acta 2012, 1823, 125–129. [Google Scholar] [CrossRef]
- Schuberth, C.; Buchberger, A. UBX domain proteins: Major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. CMLS 2008, 65, 2360–2371. [Google Scholar] [CrossRef]
- Sá-Moura, B.; Funakoshi, M.; Tomko, R.J.; Dohmen, R.J.; Wu, Z.; Peng, J.; Hochstrasser, M. A Conserved Protein with AN1 Zinc Finger and Ubiquitin-like Domains Modulates Cdc48 (p97) Function in the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 2013, 288, 33682–33696. [Google Scholar] [CrossRef]
- Hanna, J.; Waterman, D.; Isasa, M.; Elsasser, S.; Shi, Y.; Gygi, S.; Finley, D. Cuz1/Ynl155w, a zinc-dependent ubiquitin-binding protein, protects cells from metalloid-induced proteotoxicity. J. Biol. Chem. 2014, 289, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, R.; Rodrigues, J.I.; Litwin, I.; Tamás, M.J. Mechanisms of genotoxicity and proteotoxicity induced by the metalloids arsenic and antimony. Cell. Mol. Life Sci. 2023, 80, 342. [Google Scholar] [CrossRef] [PubMed]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Böhm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol. Cell 2018, 70, 906–919.e7. [Google Scholar] [CrossRef]
- Kolawa, N.; Sweredoski, M.J.; Graham, R.L.J.; Oania, R.; Hess, S.; Deshaies, R.J. Perturbations to the Ubiquitin Conjugate Proteome in Yeast Δubx Mutants Identify Ubx2 as a Regulator of Membrane Lipid Composition. Mol. Cell. Proteom. MCP 2013, 12, 2791–2803. [Google Scholar] [CrossRef]
- LaLonde, D.P.; Bretscher, A. The UBX protein SAKS1 negatively regulates endoplasmic reticulum-associated degradation and p97-dependent degradation. J. Biol. Chem. 2011, 286, 4892–4901. [Google Scholar] [CrossRef]
- Rossio, V.; Liu, X.; Paulo, J.A. Comparative proteomic analysis of two commonly used laboratory yeast strains: W303 and BY4742. Proteomes 2023, 11, 30. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, X.; Rossio, V.; Dawson, S.L.; Gygi, S.P.; Paulo, J.A. Enhancing Proteome Coverage by Using Strong Anion-Exchange in Tandem with Basic-pH Reversed-Phase Chromatography for Sample Multiplexing-Based Proteomics. J. Proteome Res. 2023, 23, 2870–2881. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Rossio, V.; Paulo, J.A.; Liu, X.; Gygi, S.P.; King, R.W. Specificity profiling of deubiquitylases against endogenously generated ubiquitin-protein conjugates. Cell Chem. Biol. 2024, 31, 1349–1362.e5. [Google Scholar] [CrossRef]
- Rossio, V.; Paulo, J.A. Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies. Proteomes 2023, 11, 28. [Google Scholar] [CrossRef]
- Liu, X.; Rossio, V.; Paulo, J.A. Spin column-based peptide fractionation alternatives for streamlined tandem mass tag (SL-TMT) sample processing. J. Proteom. 2023, 276, 104839. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Bonneil, E.; Pomiès, C.; Eng, K.; Thibault, P. Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. J. Proteome Res. 2009, 8, 3355–3366. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Villén, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-Decoy Search Strategy for Mass Spectrometry-Based Proteomics. In Proteome Bioinformatics; Hubbard, S.J., Jones, A.R., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 55–71. [Google Scholar] [CrossRef]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Paulo, J.A.; O’Connell, J.D.; Gygi, S.P. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 2016, 27, 1620–1625. [Google Scholar] [CrossRef]
- Decottignies, A.; Evain, A.; Ghislain, M. Binding of Cdc48p to a ubiquitin-related UBX domain from novel yeast proteins involved in intracellular proteolysis and sporulation. Yeast 2004, 21, 127–139. [Google Scholar] [CrossRef]
- Mannhaupt, G.; Schnall, R.; Karpov, V.; Vetter, I.; Feldmann, H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 1999, 450, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, R.J.; Willers, I.; Marques, A.J. Biting the hand that feeds: Rpn4-dependent feedback regulation of proteasome function. Biochim. Biophys. Acta 2007, 1773, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Moreno, A.; Isasa, M.; Bhanu, M.K.; Waterman, D.P.; Eapen, V.V.; Gygi, S.P.; Hanna, J. Proteomic Analysis Identifies Ribosome Reduction as an Effective Proteotoxic Stress Response. J. Biol. Chem. 2015, 290, 29695–29706. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, C.; Richly, H.; Rumpf, S.; Buchberger, A. Shp1 and Ubx2 are adaptors of Cdc48 involved in ubiquitin-dependent protein degradation. EMBO Rep. 2004, 5, 818–824. [Google Scholar] [CrossRef]
- Li, H.; Ji, Z.; Paulo, J.A.; Gygi, S.P.; Rapoport, T.A. Bidirectional substrate shuttling between the 26S proteasome and the Cdc48 ATPase promotes protein degradation. Mol. Cell 2024, 84, 1290–1303.e7. [Google Scholar] [CrossRef]
- Chowdhury, A.; Ogura, T.; Esaki, M. Two Cdc48 cofactors Ubp3 and Ubx2 regulate mitochondrial morphology and protein turnover. J. Biochem. 2018, 164, 349–358. [Google Scholar] [CrossRef]
- Simões, T.; Schuster, R.; den Brave, F.; Escobar-Henriques, M. Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. eLife 2018, 7, e30015. [Google Scholar] [CrossRef]
- Esaki, M.; Ogura, T. Cdc48p/p97-mediated regulation of mitochondrial morphology is Vms1p-independent. J. Struct. Biol. 2012, 179, 112–120. [Google Scholar] [CrossRef]
- Chen, Y.; Sanchez, Y. Chk1 in the DNA damage response: Conserved roles from yeasts to mammals. DNA Repair. 2004, 3, 1025–1032. [Google Scholar] [CrossRef]
- Kaps, S.; Kettner, K.; Migotti, R.; Kanashova, T.; Krause, U.; Rödel, G.; Dittmar, G.; Kriegel, T.M. Protein kinase Ymr291w/Tda1 is essential for glucose signaling in saccharomyces cerevisiae on the level of hexokinase isoenzyme ScHxk2 phosphorylation. J. Biol. Chem. 2015, 290, 6243–6255. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossio, V.; Paulo, J.A. Comparative Proteome-Wide Abundance Profiling of Yeast Strains Deleted for Cdc48 Adaptors. Proteomes 2024, 12, 31. https://doi.org/10.3390/proteomes12040031
Rossio V, Paulo JA. Comparative Proteome-Wide Abundance Profiling of Yeast Strains Deleted for Cdc48 Adaptors. Proteomes. 2024; 12(4):31. https://doi.org/10.3390/proteomes12040031
Chicago/Turabian StyleRossio, Valentina, and Joao A. Paulo. 2024. "Comparative Proteome-Wide Abundance Profiling of Yeast Strains Deleted for Cdc48 Adaptors" Proteomes 12, no. 4: 31. https://doi.org/10.3390/proteomes12040031
APA StyleRossio, V., & Paulo, J. A. (2024). Comparative Proteome-Wide Abundance Profiling of Yeast Strains Deleted for Cdc48 Adaptors. Proteomes, 12(4), 31. https://doi.org/10.3390/proteomes12040031