
Pattern Analysis of Organellar Maps for Interpretation of Proteomic Data

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
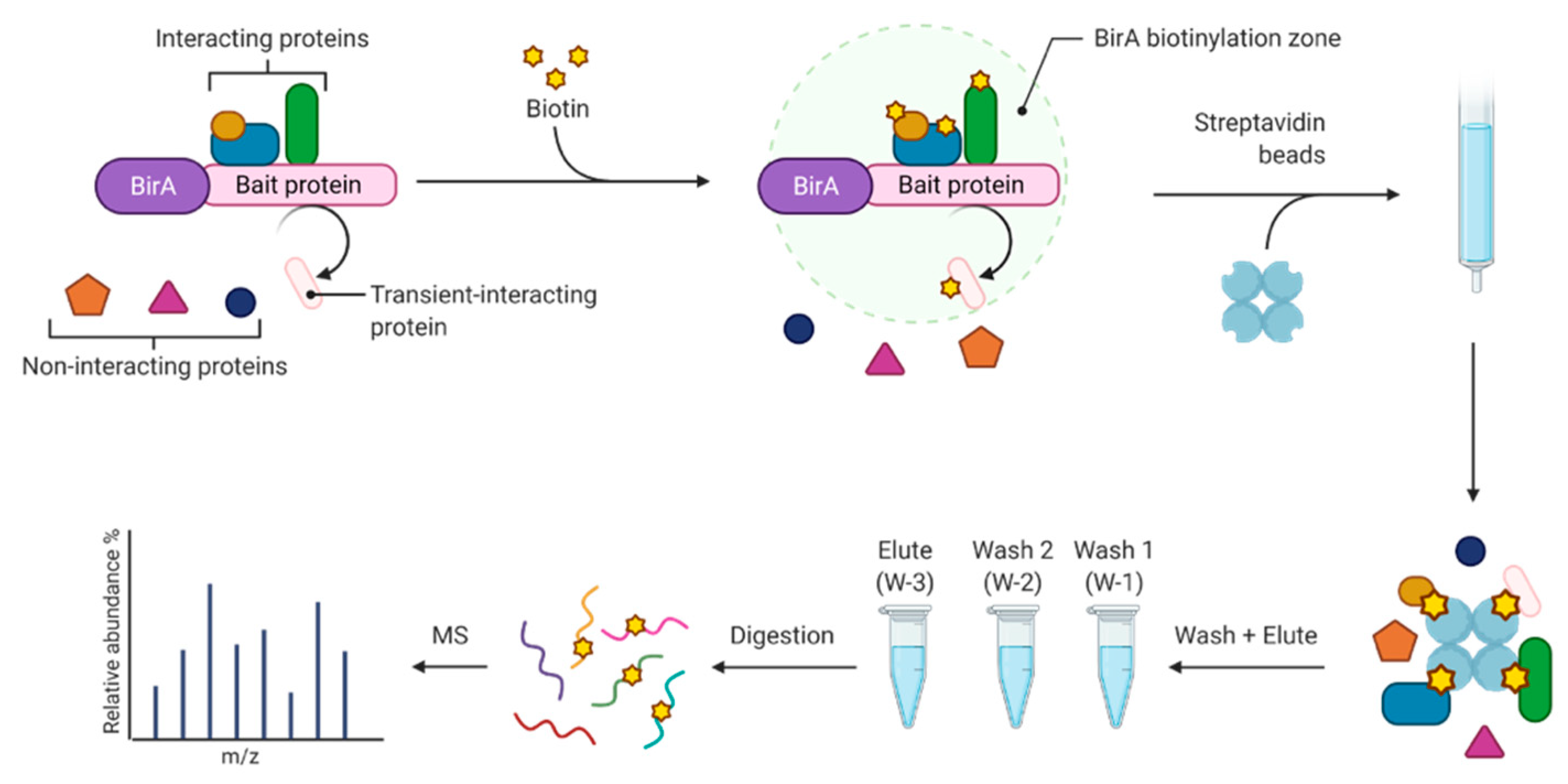
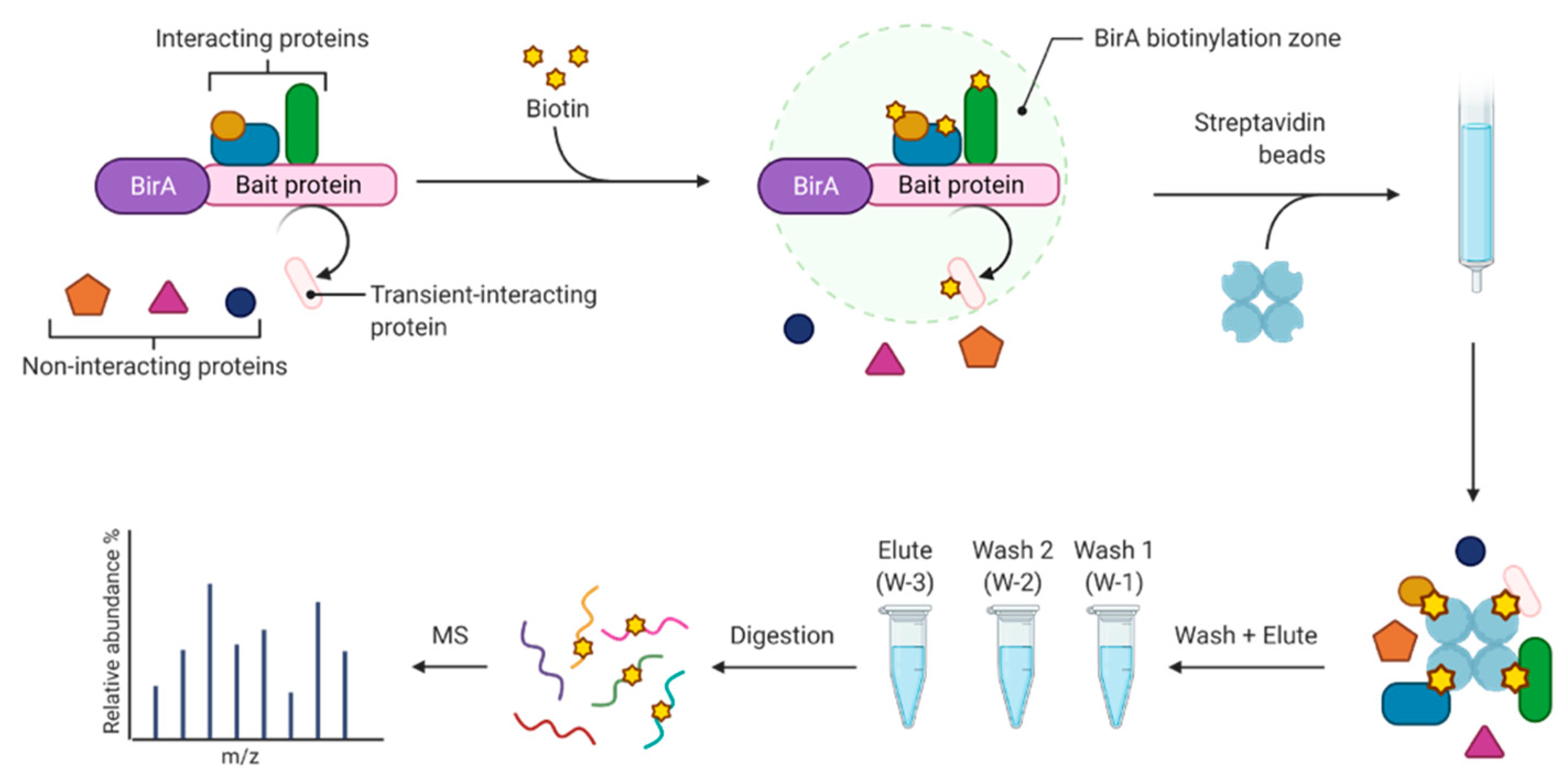
2.1. Biotin Affinity Selection
2.1.1. Biotin Proximity Labeling Workflow
2.1.2. Liquid Chromatography Mass Spectrometry
2.1.3. Database Searching
2.2. Publicly Available Data
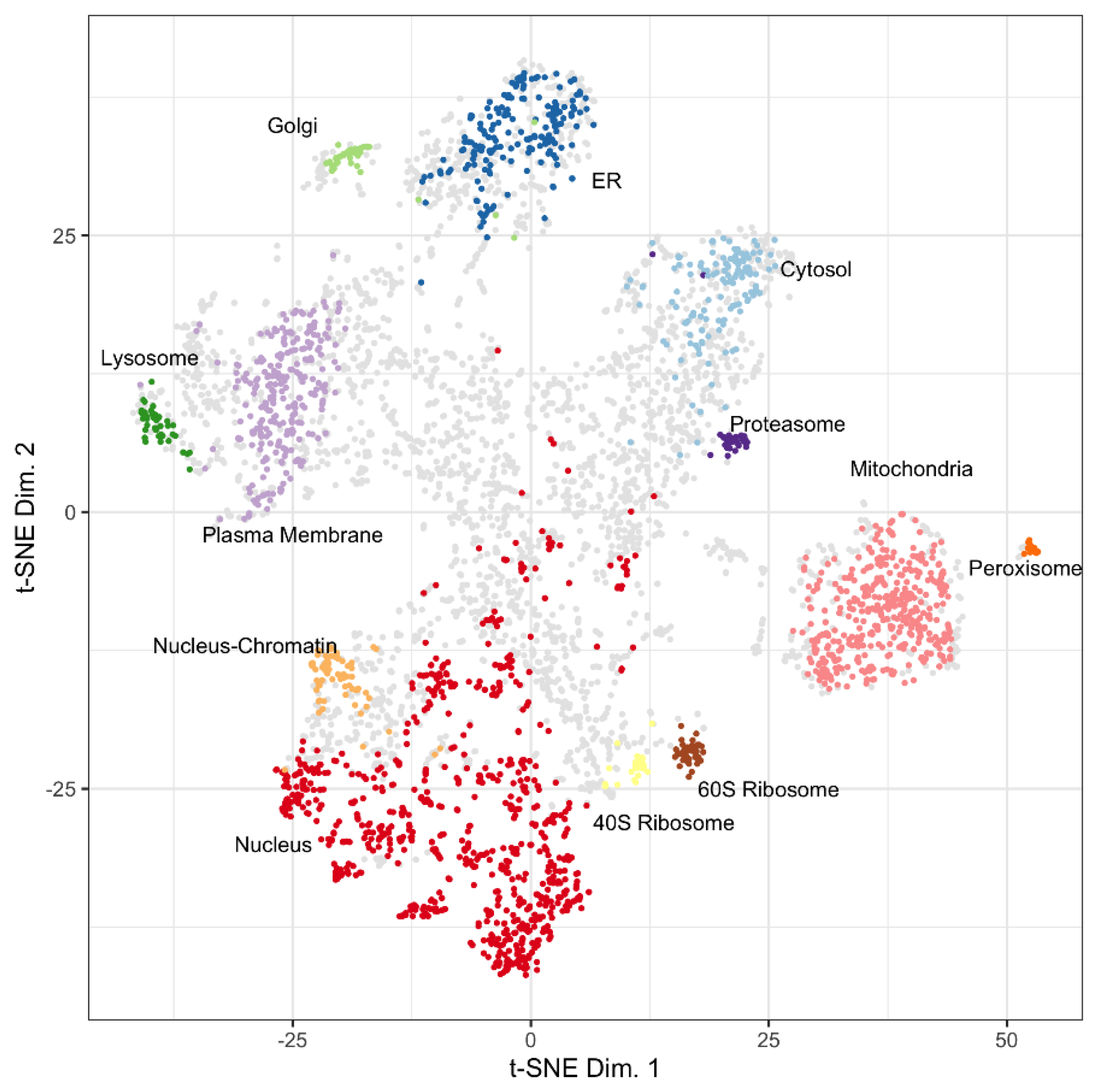
2.2.1. LOPIT Data
2.2.2. Exosome Enrichment by Density Gradient Ultracentrifugation
2.2.3. Exosome Enrichment by Size Exclusion Chromatography, Density Gradient Ultracentrifugation, and Ultracentrifugation
2.3. Data Processing, Visualization, and Availability
3. Results
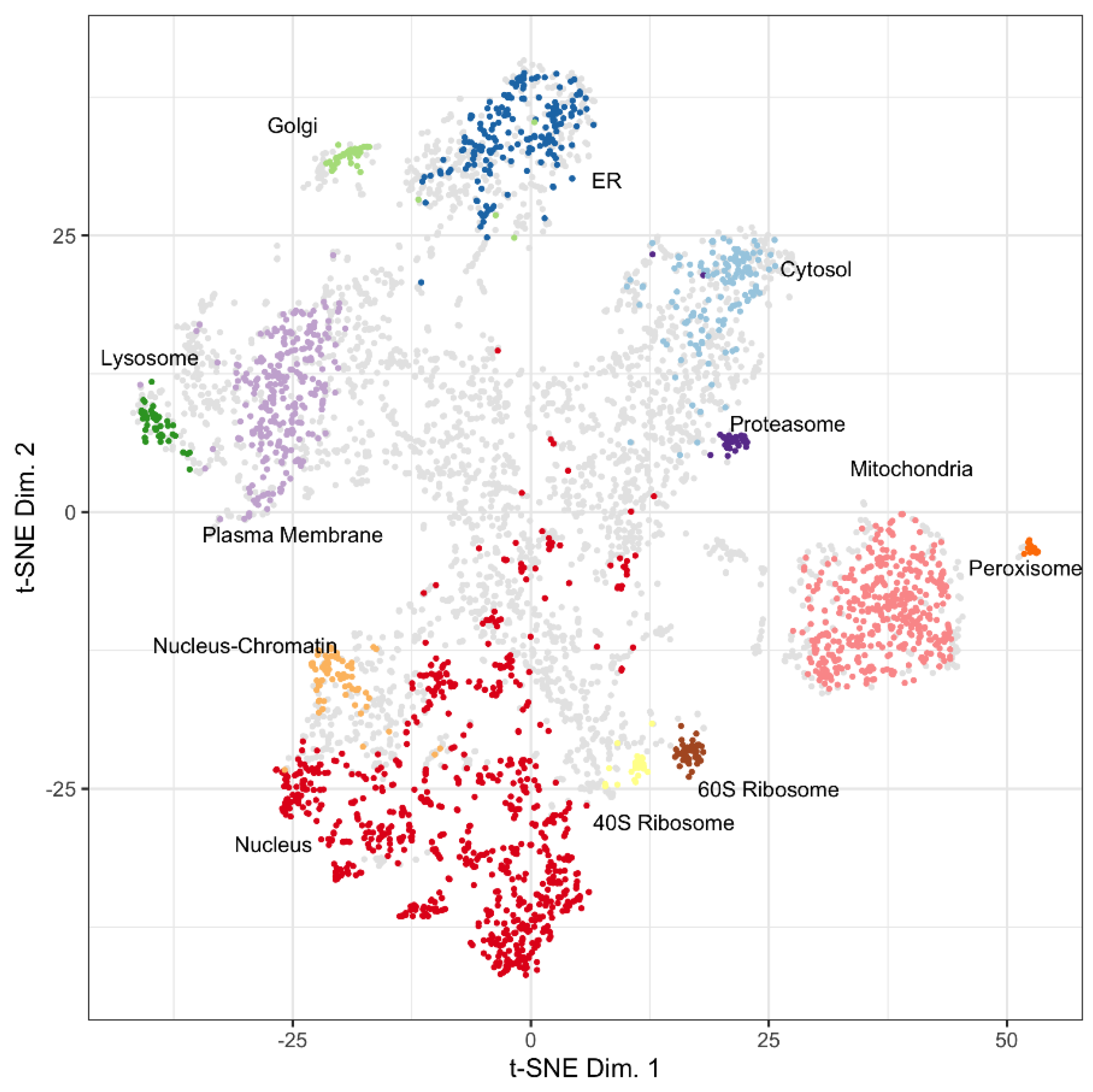
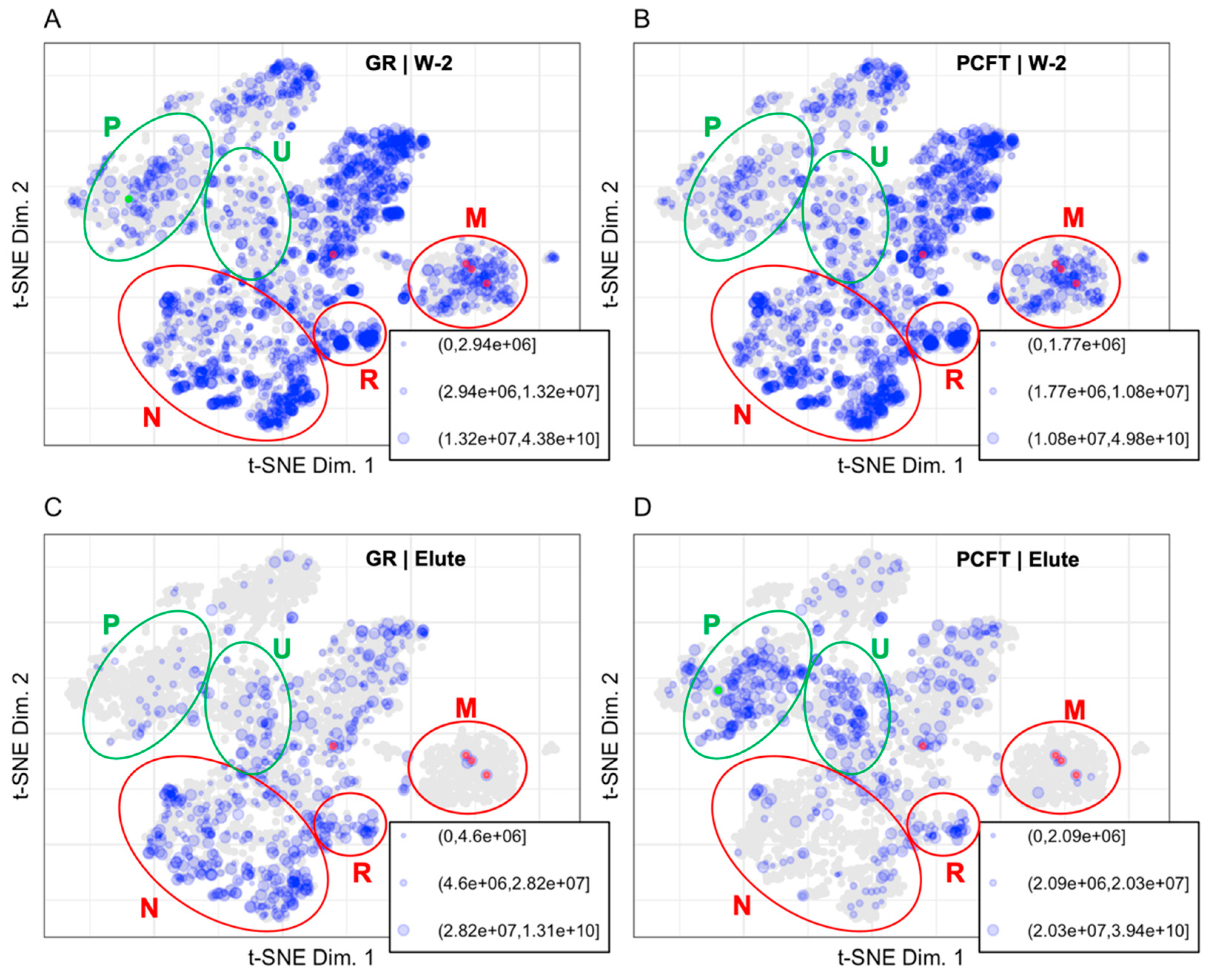
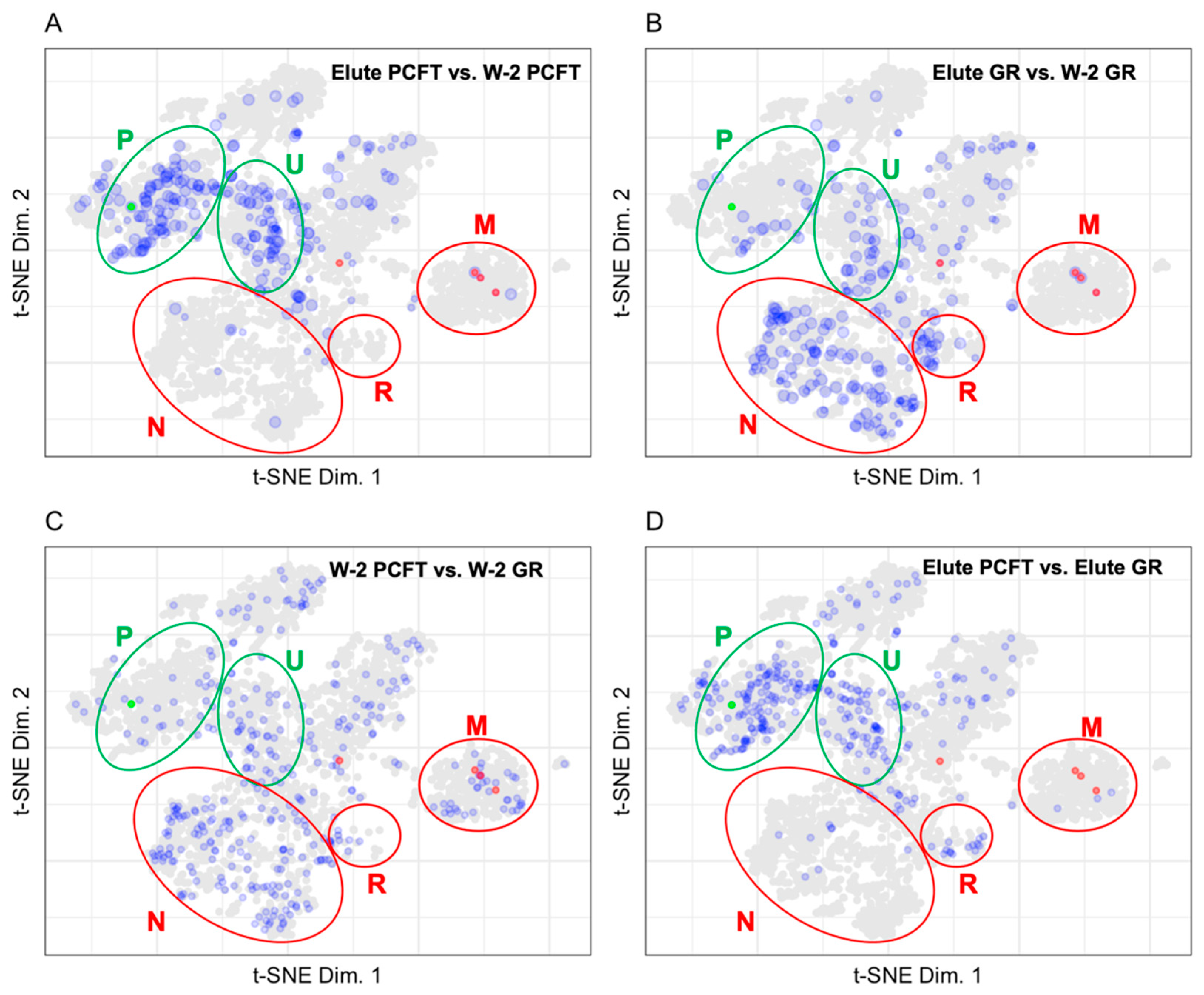
3.1. Mapping Proteins from a Proximity Labeling Experiment to the LOPIT Plot
3.1.1. Protein Abundances
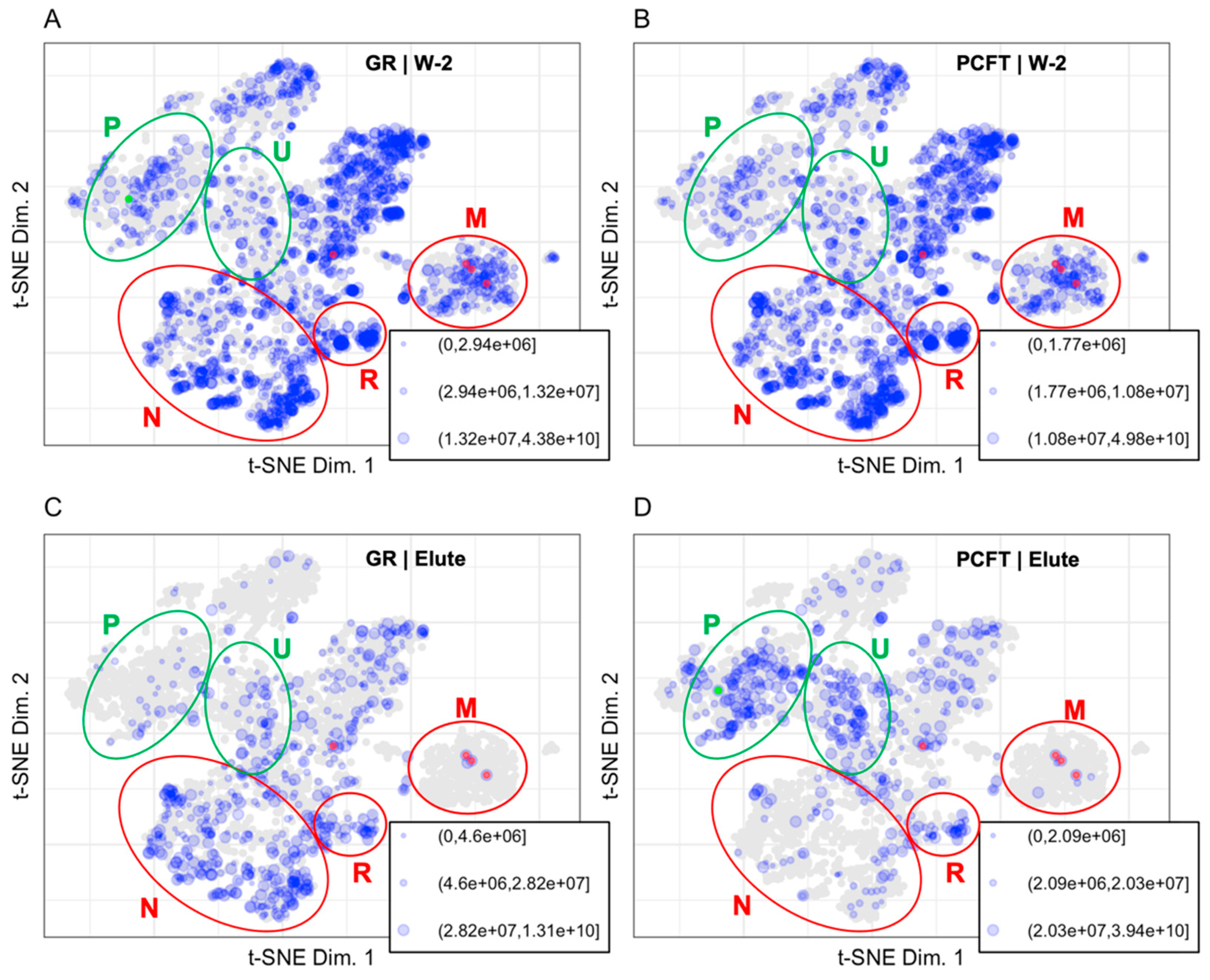
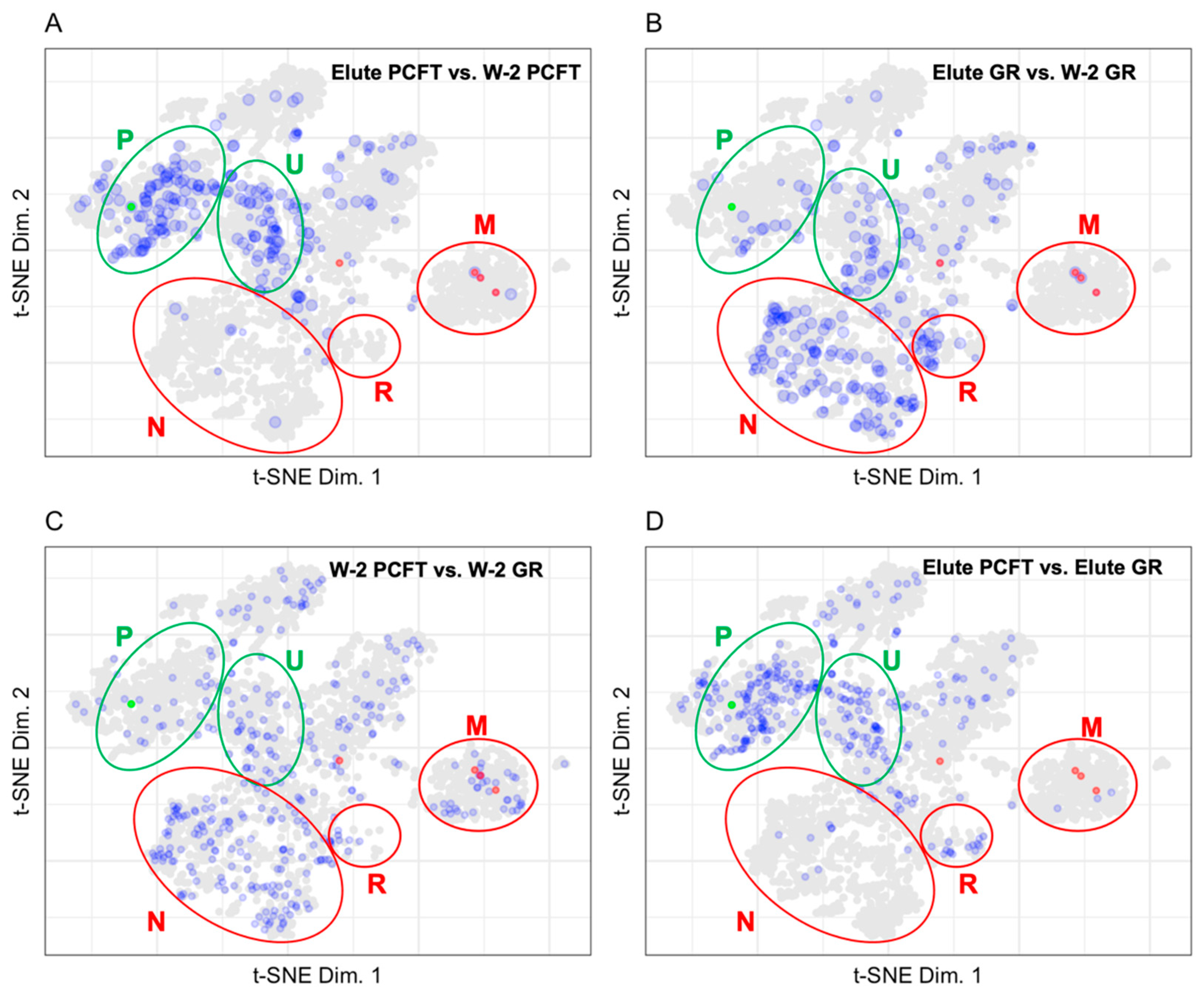
3.1.2. Mapping BioID Wash to Elute Ratio Data
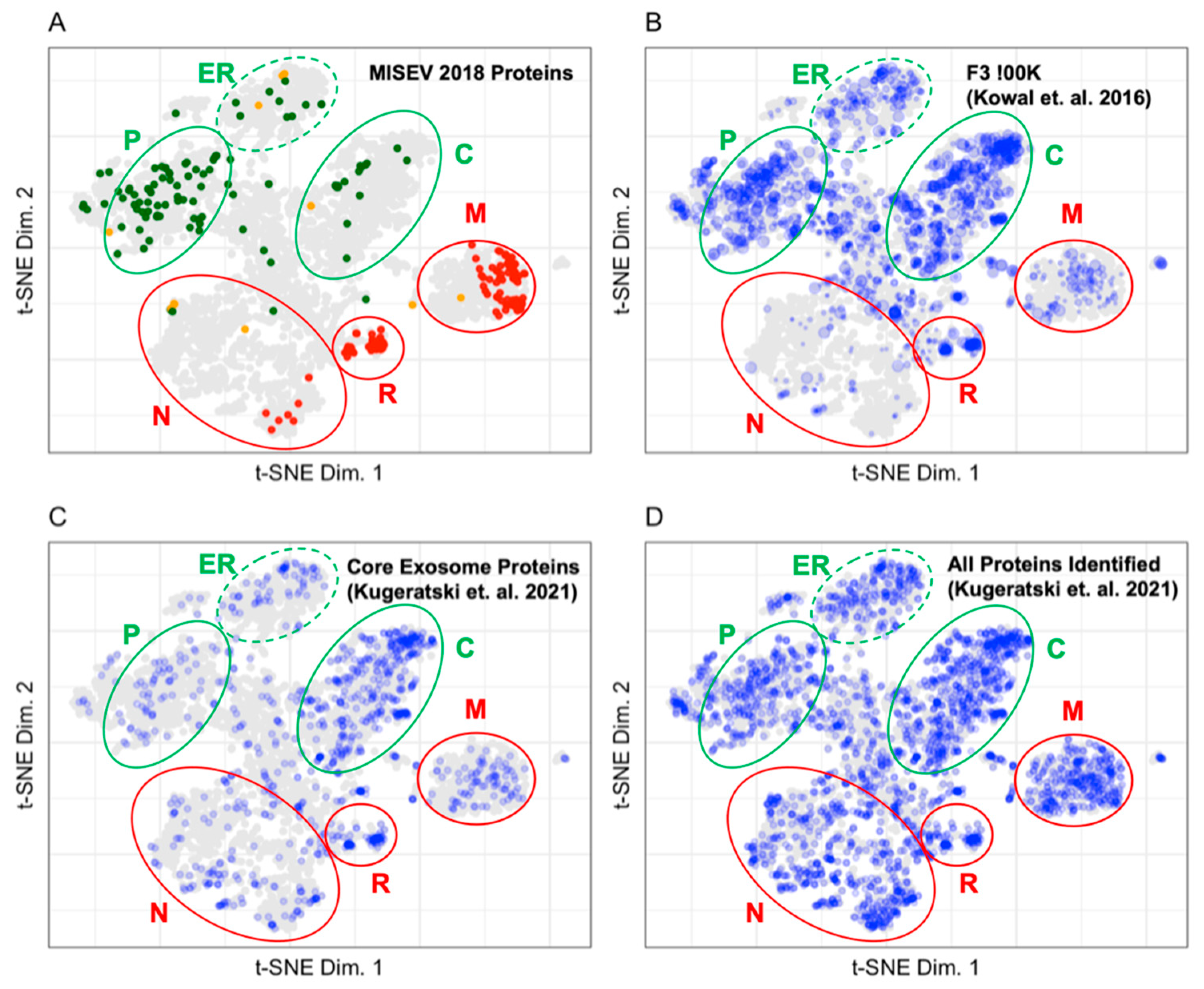
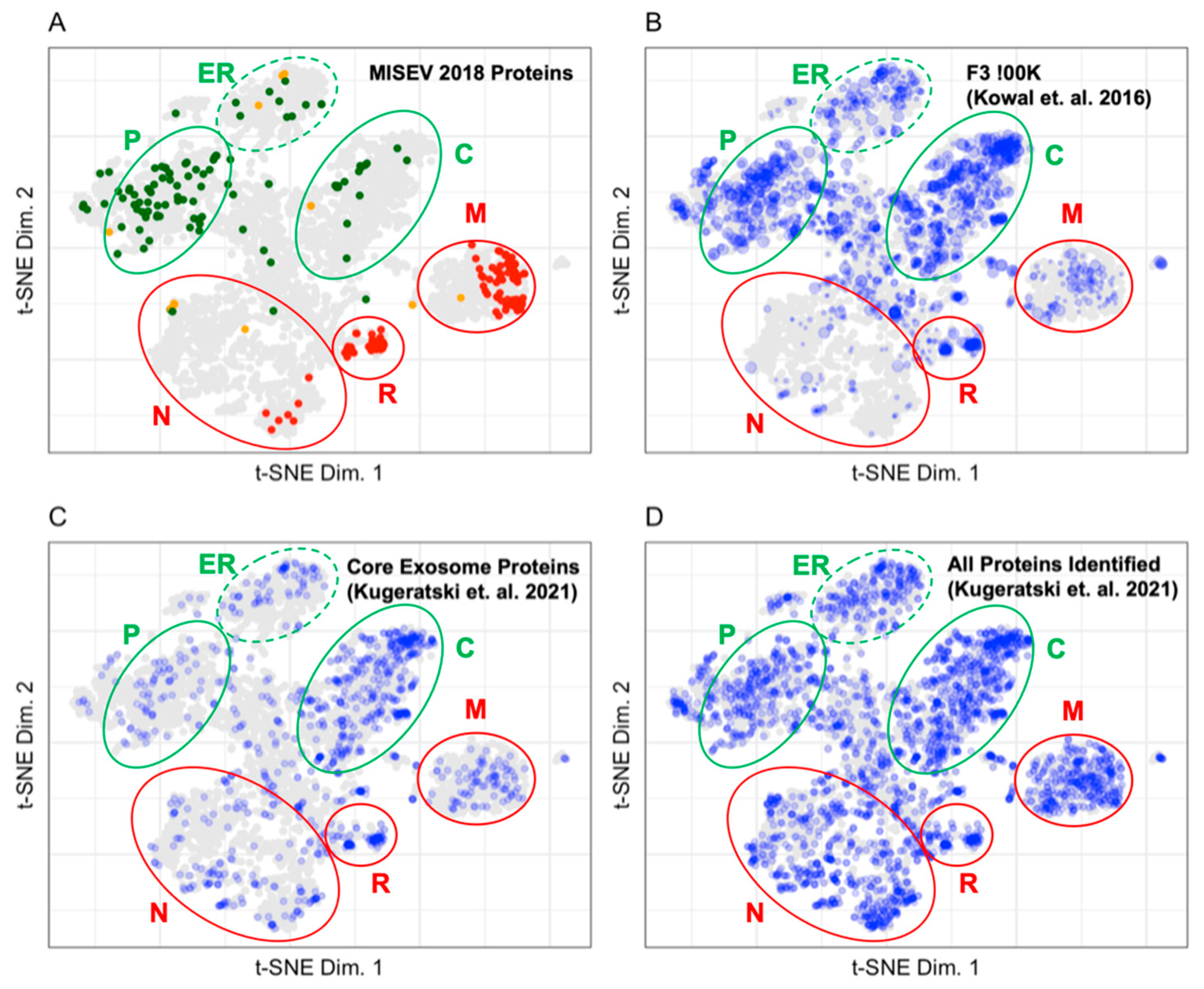
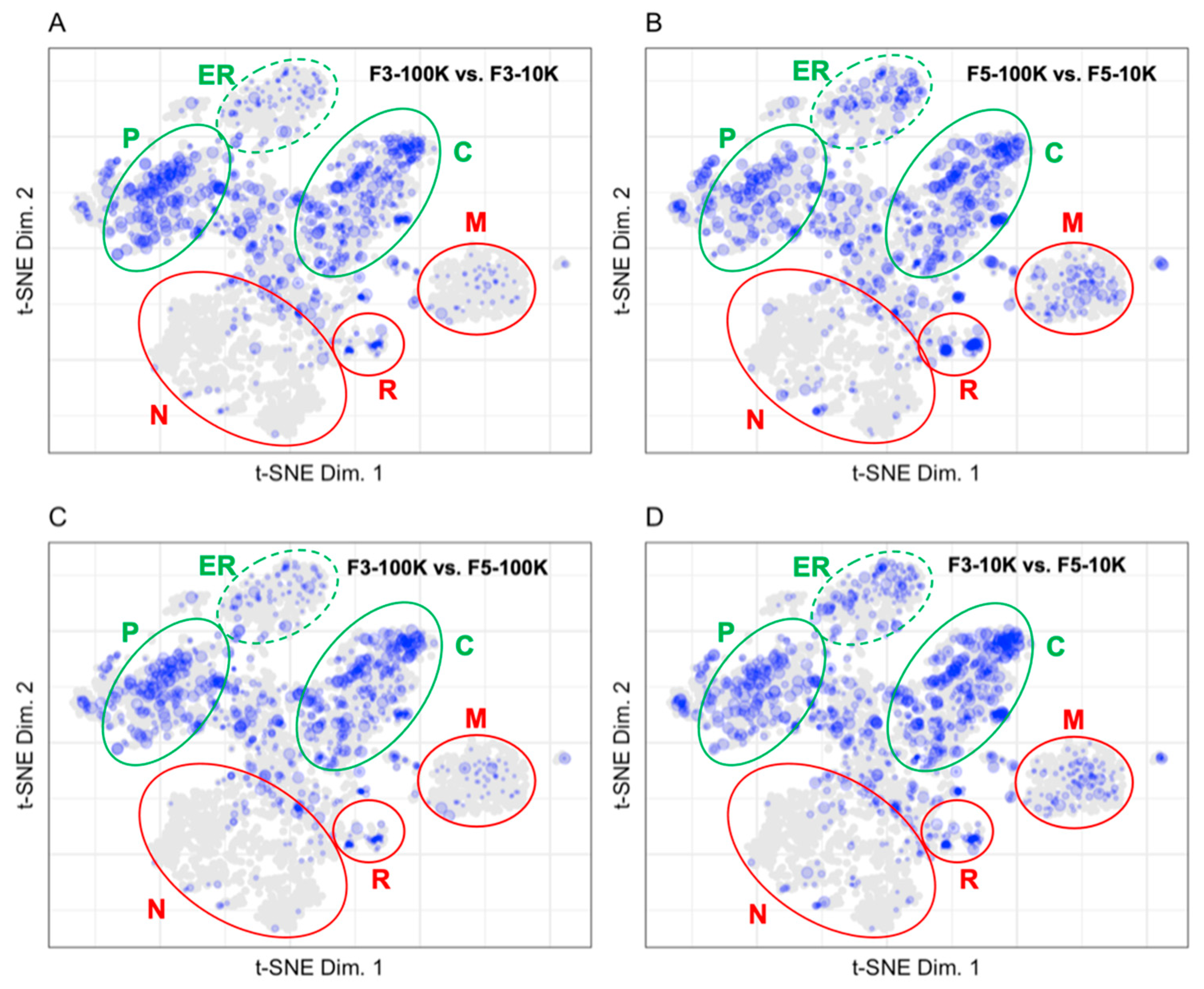
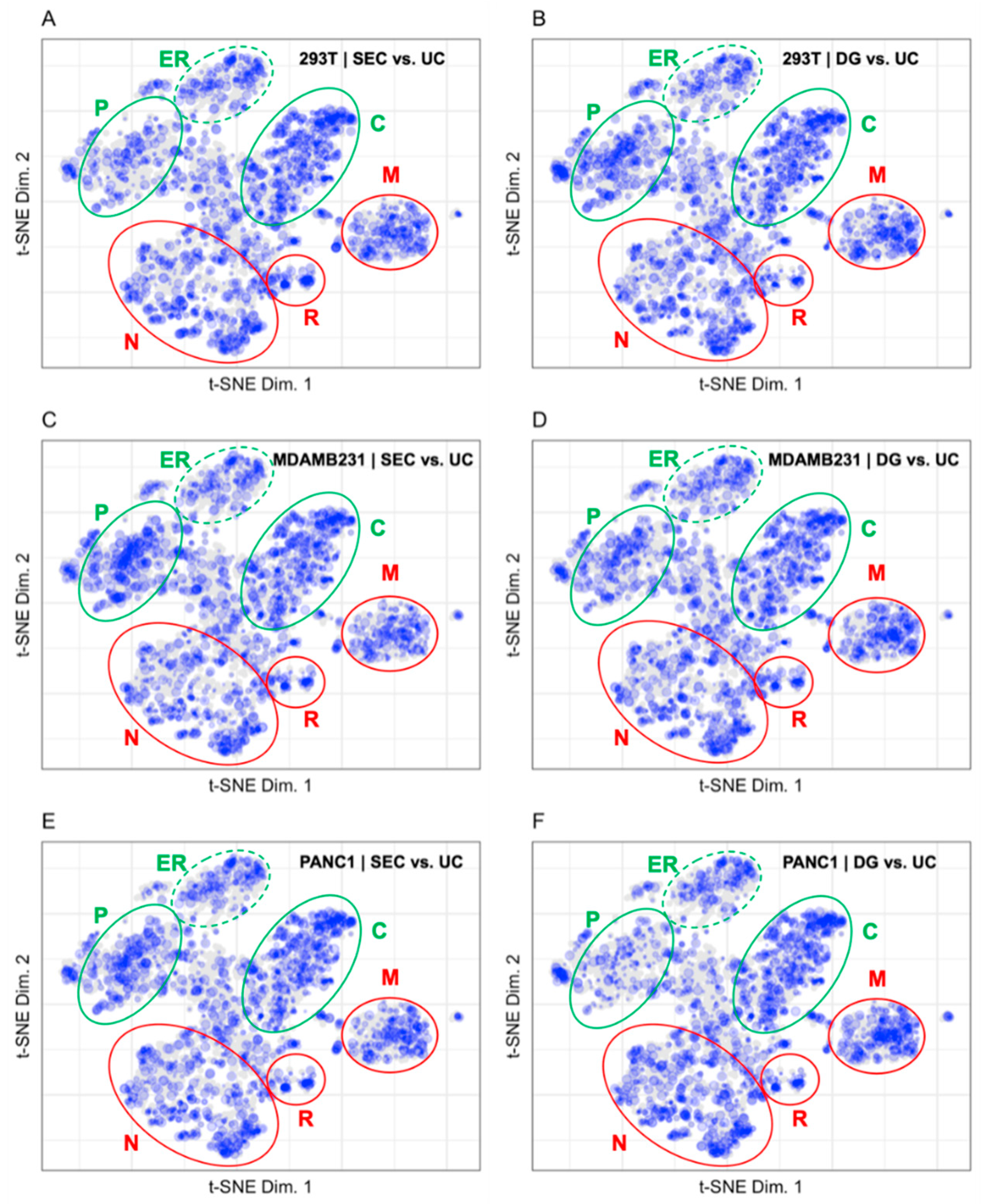
3.2. Mapping Proteins from Extracellular Vesicle Preparations to the LOPIT Plot
4. Discussion
4.1. LOPIT Plots for the Interpretation of Data from a BioID Proximity Labeling Experiment
4.2. Pattern Recognition Is Useful for the Interpretation of Data from Extracellular Vesicle Preparations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pankow, S.; Martínez-Bartolomé, S.; Bamberger, C.; Yates, J.R. Understanding molecular mechanisms of disease through spatial proteomics. Curr. Opin. Chem. Biol. 2019, 48, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, T.P.J.; Watson, R.; Griffin, J.L.; Dupree, P.; Lilley, K.S. Localization of Organelle Proteins by Isotope Tagging (LOPIT). Mol. Cell. Proteom. 2004, 3, 1128–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; At Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Geladaki, A.; Britovsek, N.K.; Breckels, L.M.; Smith, T.S.; Vennard, O.L.; Mulvey, C.M.; Crook, O.M.; Gatto, L.; Lilley, K.S. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 2019, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, C.M.; Breckels, L.M.; Geladaki, A.; Britovesk, N.K.; Nightingale, D.J.H.; Chrisoforou, A.; Elzek, M.; Deery, M.J.; Gatto, L.; Lilley, K.S. Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat. Protoc. 2017, 12, 1110–1135. [Google Scholar] [CrossRef]
- Gatto, L.; Breckels, L.M.; Wieczorek, S.; Burger, T.; Lilley, K.S. Mass-spectometry-based spatial proteomics data analysis using pRoloc and pRolocdata. Bioinformatics 2014, 30, 1322–1324. [Google Scholar] [CrossRef] [Green Version]
- Grimes, M.L.; Lee, W.J.; Van Der Maaten, L.; Shannon, P. Wrangling Phosphoproteomic Data to Elucidate Cancer Signaling Pathways. PLoS ONE 2013, 8, e52884. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Go, C.D.; Kinght, J.D.R.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.Y.; Samavarchi-Tehrani, P.; Zhang, H.; Zhu, L.Y.; et al. A proximity-dependent biotinylation map of a human cell. Nature 2021, 595, 120–124. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Samson, R.; Gingras, A.C. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell. Proteom. 2020, 19, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndres, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.B.; Carruthers, N.J.; Stemmer, P.M. Enriching extracellular vesicles for mass spectrometry. Mass Spectrom. Rev. 2021. [CrossRef]
- Whittaker, T.E.; Nagelkerke, A.; Nele, V.; Kausher, U.; Stevens, M.M. Experimental artefacts can lead to misattribution of bioactivity from soluble mesenchymal stem cell paracrine factors to extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1807674. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Goldman, I.D. The molecular identity and characterization of a proton-coupled folate transporter--PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer Metast. Rev. 2007, 26, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Gangjee, A.; Matherly, L.H. The evolving biology of the proton-coupled folate transporter: New insights into regulation, structure, and mechanism. Faseb J. 2022, 36, e22164. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Nobel, K.A.; Birendra, K.C.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Holmberg, A.; Eidefors, A.; Nord, O.; Lukacs, M.; Lundeberg, J.; Uhlen, M. The biotin-Streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 2005, 26, 501–510. [Google Scholar] [CrossRef]
- Tytgat, H.L.P.; Schoofs, G.; Driesen, M.; Proost, P.; Van Damme, E.J.M.; Vanderleyden, J.; Lebeer, S. Endogenous biotin-binding proteins: An overlooked factor causing false positives in streptavidin-based protein detection. Microb. Biotechnol. 2015, 8, 164–168. [Google Scholar] [CrossRef]
- Nakai, Y.; Inoue, K.; Abe, N.; Hatakeyama, M.; Ohta, K.; Otagiri, M.; Hayashi, Y.; Yuasa, H. Functional Characterization of Human Proton-Coupled Folate Transporter/Heme Carrier Protein 1 Heterologously Expressed in Mammalian Cells as a Folate Transporter. J. Pharmacol. Exp. Ther. 2007, 322, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royo, F.; Thery, C.; Falcon-Perez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for separation and characterization of extracellular vesicles: Results of a worldwide survey performed by the ISEV rigor and standardization subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Total Protein IDs | Total LOPIT Proteins | GR Protein IDs | GR LOPIT Proteins (%) | PCFT Protein IDs | PCFT LOPIT Proteins (%) |
|---|---|---|---|---|---|---|
| W-1 | 2330 | 1986 | 2297 | 85.42% | 2284 | 85.42% |
| W-2 | 3249 | 2403 | 2375 | 79.83% | 2222 | 79.48% |
| Elute | 1220 | 887 | 781 | 75.93% | 687 | 72.78% |
| Category | Proteins | Mapped Proteins | Proteins Included | LOPIT Color |
|---|---|---|---|---|
| 1 | 110 | 38 | Proteins associated to the plasma membrane and/or endosomes | Green |
| 2 | 55 | 35 | Proteins recovered in the cytosol of extracellular vesicles | Green |
| 3 | 195 | 157 | Non-extracellular vesicle co-isolated structures | Red |
| 4 | 41 | 14 | Markers for extracellular vesicle subtypes | Orange |
| 5 | 160 | 28 | Functional component of EVs | Green |
| Assignment | MISEV 2018 Mapped Proteins | Kowal et. al. F3-100K Mapped Proteins | Kugeratski et al. Core Exosome Mapped Proteins | Kugeratski et al. Cell Exosome Mapped Proteins |
|---|---|---|---|---|
| Cytosol | 2 | 94 | 82 | 109 |
| Endoplasmic reticulum | 6 | 79 | 42 | 108 |
| Golgi | 0 | 4 | 2 | 15 |
| Lysosome | 2 | 20 | 2 | 27 |
| Mitochondria | 45 | 90 | 78 | 240 |
| Nucleus | 9 | 68 | 112 | 429 |
| Nucleus-chromatin | 8 | 3 | 10 | 35 |
| Peroxisome | 0 | 6 | 3 | 9 |
| Plasma membrane | 29 | 127 | 34 | 128 |
| Proteasome | 0 | 32 | 27 | 29 |
| Ribosome 40S | 30 | 29 | 23 | 31 |
| Ribosome 60S | 47 | 39 | 39 | 44 |
| Unknown | 94 | 995 | 454 | 1349 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burton, J.B.; Carruthers, N.J.; Hou, Z.; Matherly, L.H.; Stemmer, P.M. Pattern Analysis of Organellar Maps for Interpretation of Proteomic Data. Proteomes 2022, 10, 18. https://doi.org/10.3390/proteomes10020018
Burton JB, Carruthers NJ, Hou Z, Matherly LH, Stemmer PM. Pattern Analysis of Organellar Maps for Interpretation of Proteomic Data. Proteomes. 2022; 10(2):18. https://doi.org/10.3390/proteomes10020018
Chicago/Turabian StyleBurton, Jordan B., Nicholas J. Carruthers, Zhanjun Hou, Larry H. Matherly, and Paul M. Stemmer. 2022. "Pattern Analysis of Organellar Maps for Interpretation of Proteomic Data" Proteomes 10, no. 2: 18. https://doi.org/10.3390/proteomes10020018
APA StyleBurton, J. B., Carruthers, N. J., Hou, Z., Matherly, L. H., & Stemmer, P. M. (2022). Pattern Analysis of Organellar Maps for Interpretation of Proteomic Data. Proteomes, 10(2), 18. https://doi.org/10.3390/proteomes10020018