Serum Proteome Analysis for Profiling Predictive Protein Markers Associated with the Severity of Skin Lesions Induced by Ionizing Radiation

Abstract
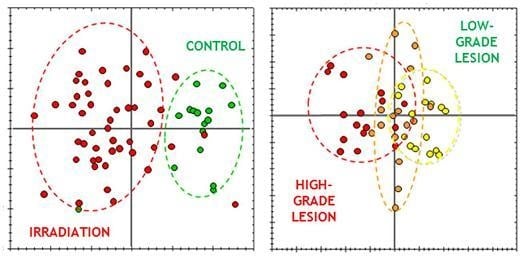
:
1. Introduction
2. Experimental
2.1. Animals
2.2. Dorsal Skin Irradiation of Mice
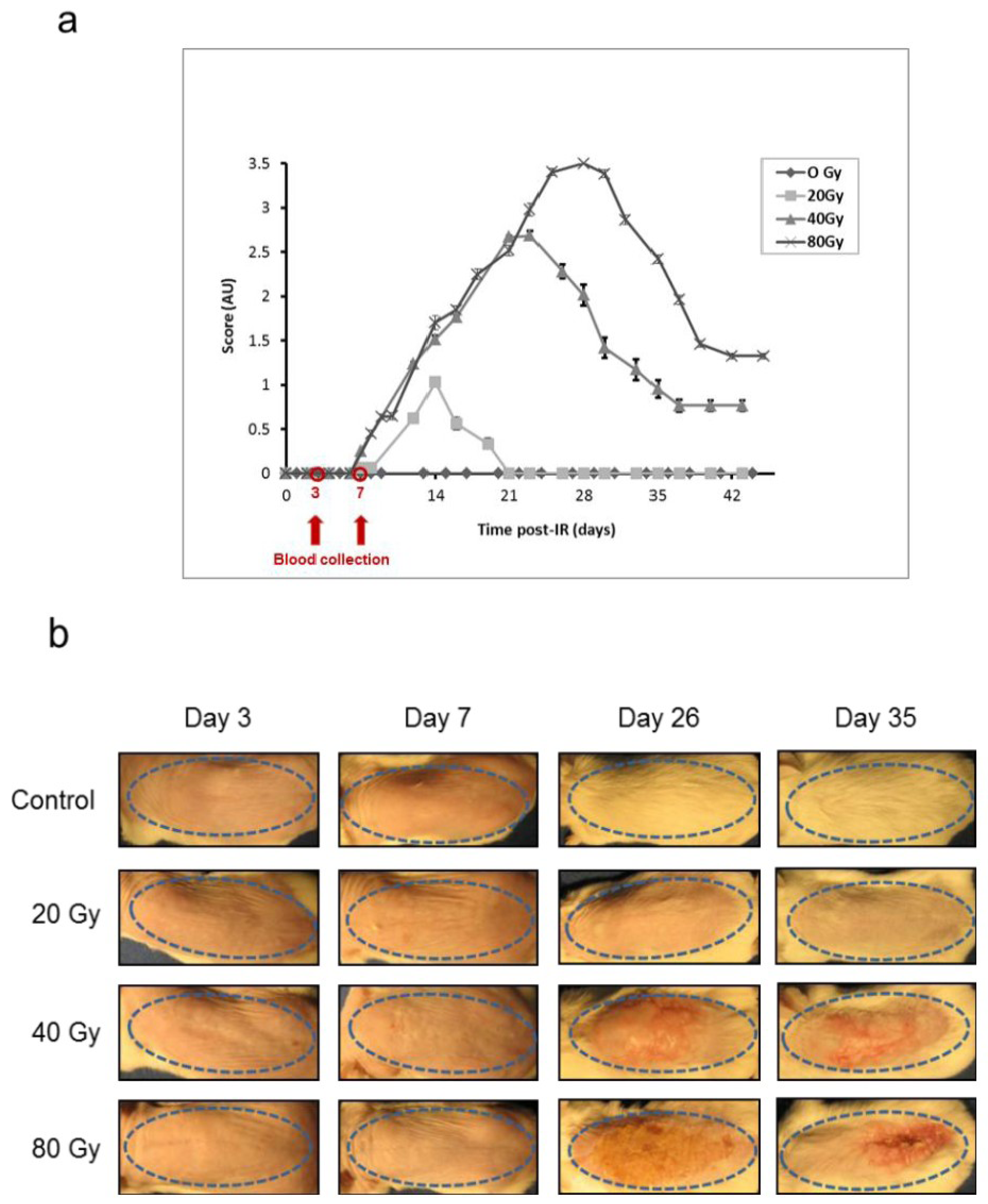
2.3. Mouse Skin Lesion Scoring System
2.4. Mouse Biological Sampling and Affinity Depletion of High Abundance Proteins
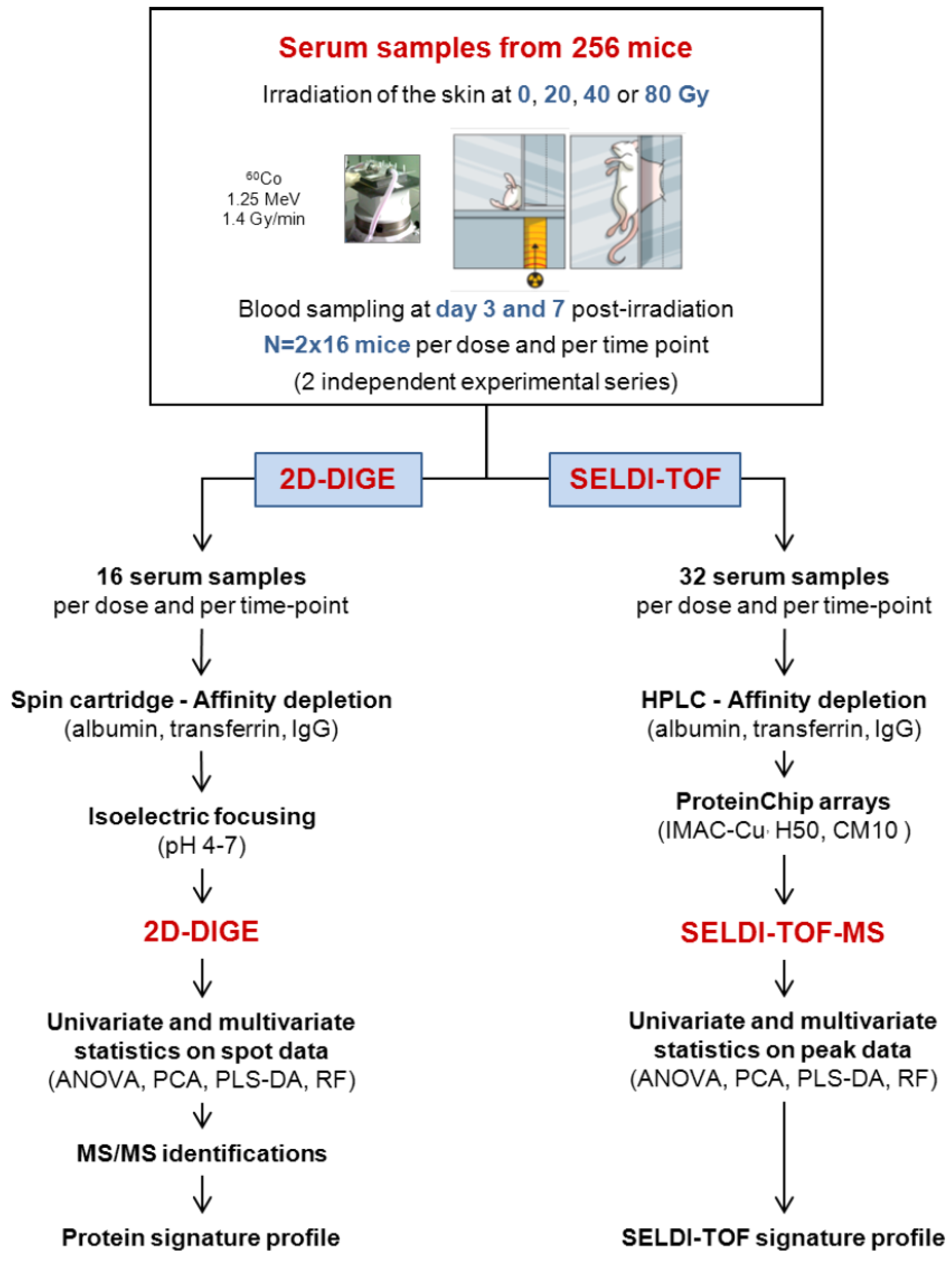
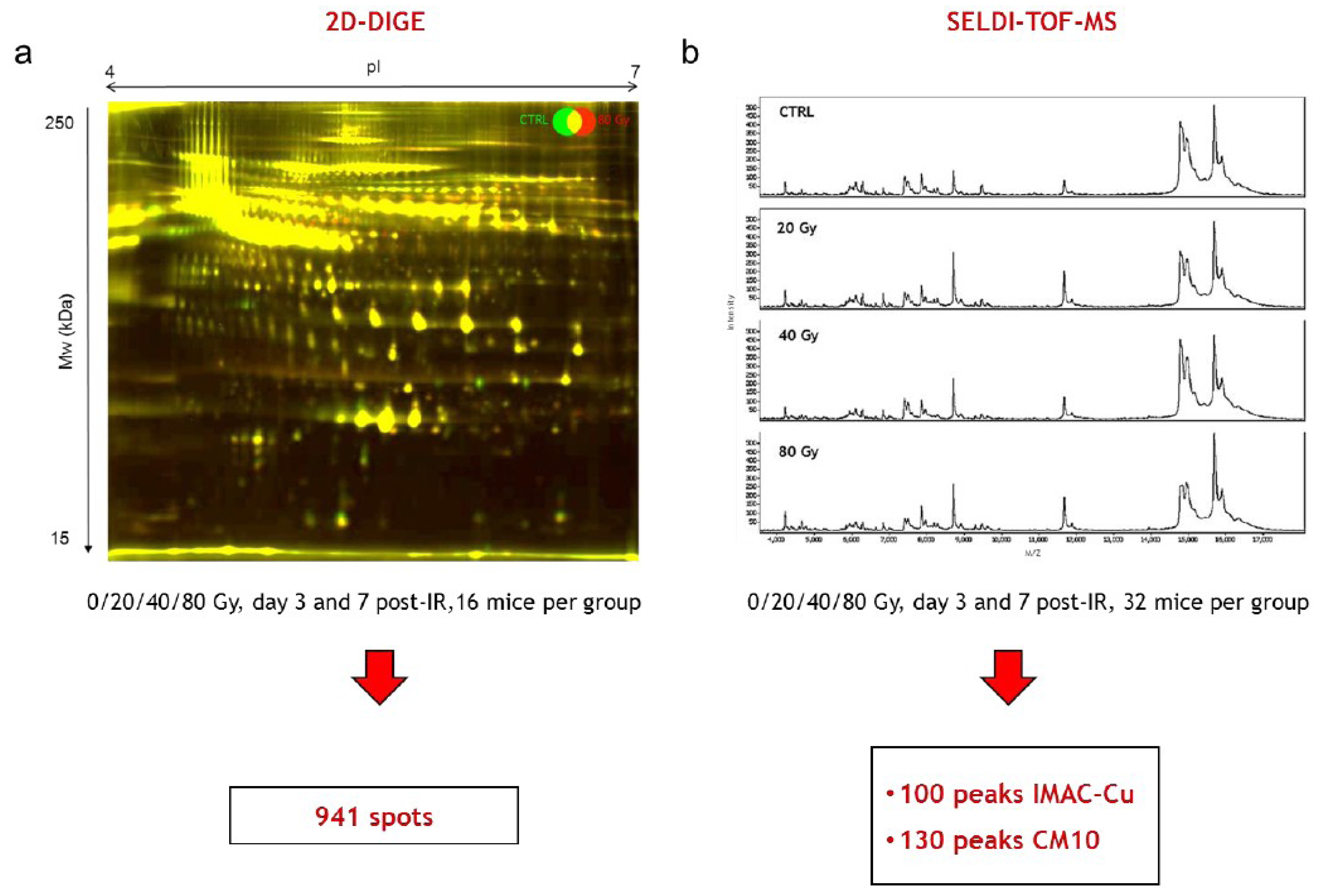
2.5. 2D-DIGE Experimental Design
2.6. Two-Dimensional Gel Electrophoresis and Imaging
2.7. 2D-DIGE Data Evaluation and Statistical Analysis
2.8. SELDI-TOF Experiments
2.9. Multivariate Statistical Analysis
2.10. Protein Identification
2.11. Pathway Analysis of Differentially Expressed Proteins
3. Results and Discussion
3.1. Experimental Strategy
3.2. Animal Model and Scoring of Irradiated Mouse Skin Lesions
3.3. Differential 2D-DIGE and SELDI-TOF Analyses of Serum Proteins


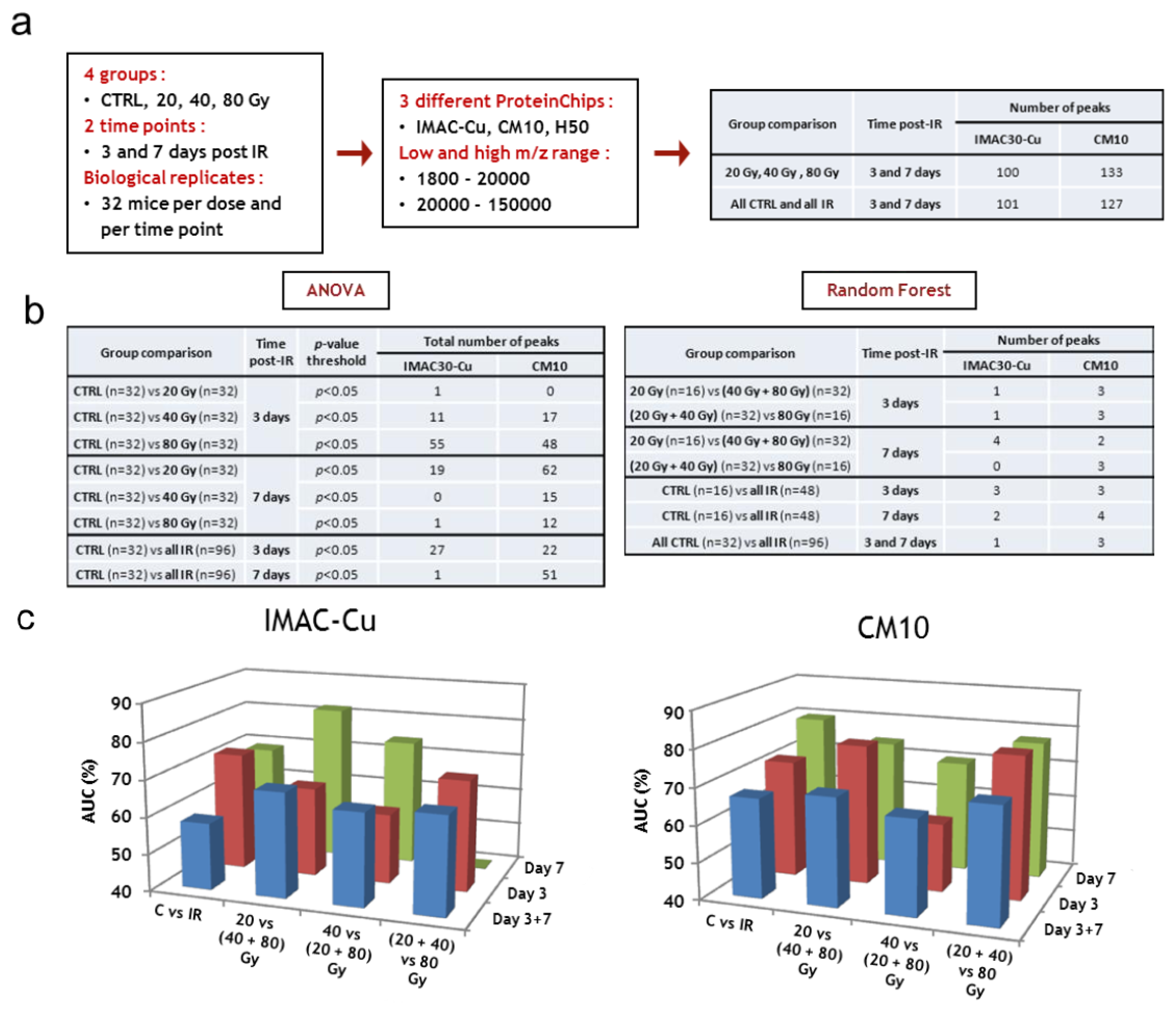
3.3.1. SELDI-TOF Analysis
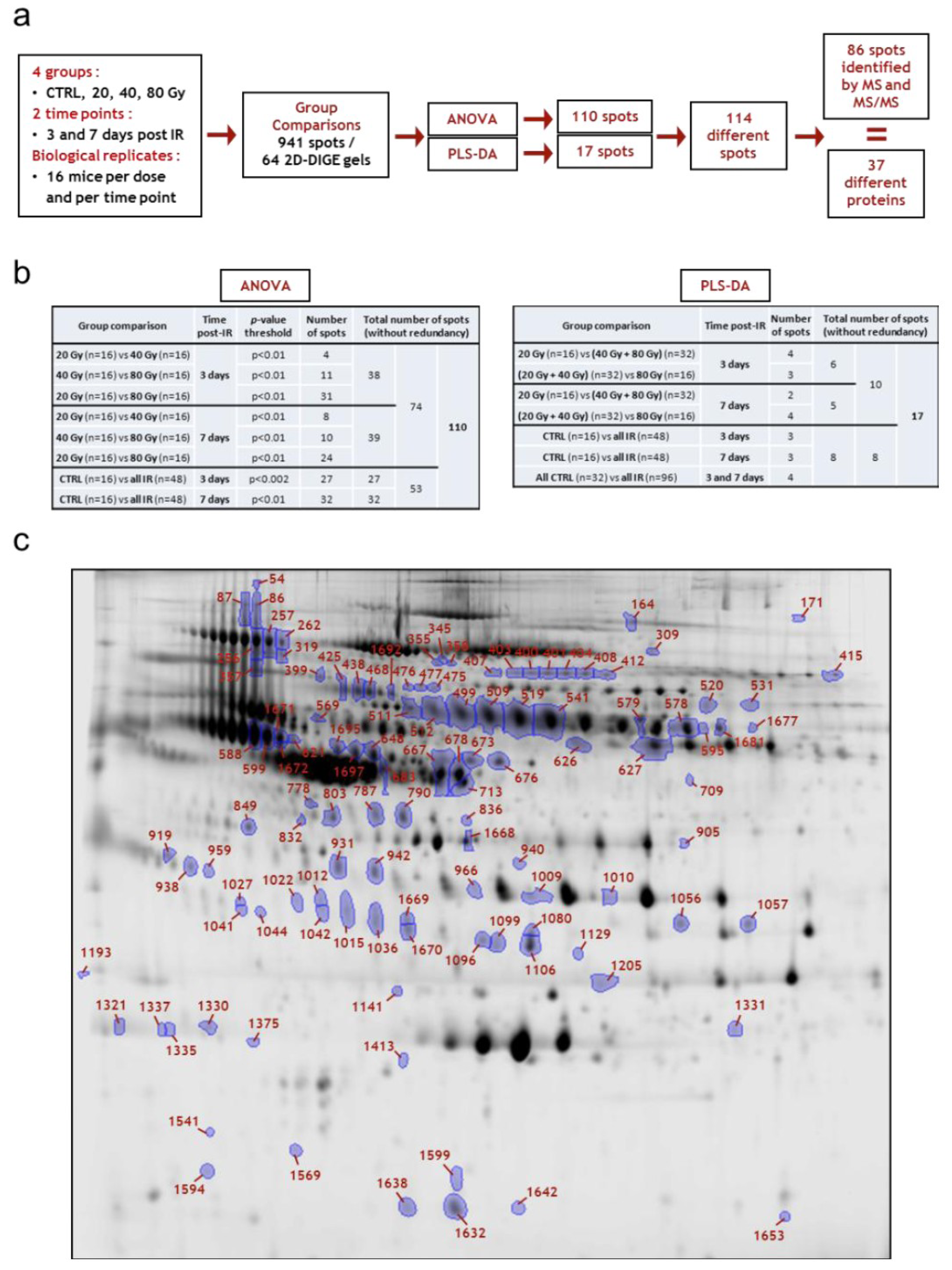
3.3.2. Differential 2D-DIGE Analysis and MS Identifications
3.3.3. Pathway Analysis of Differentially Expressed Proteins Revealed by 2D-DIGE Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein name | Name in database | SwissProt ID | Number of spots 1 | Spot number 2 | Irradiation 3,4 | Dose 3,5 | ||
|---|---|---|---|---|---|---|---|---|
| Max fold change (p < 0.05) | p-value | Max fold change (p < 0.05) | p-value | |||||
| Actin, beta | ACTB | ACTB_MOUSE | 1 | 1668 | = | / | −1.2 | 8.5 10−3 |
| Afamin | AFM | AFAM_MOUSE | 3 | 425, 438, 468 | +1.2 | 5.4 10−4 | +1.2 | 5.3 10−3 |
| Alpha-1-acid glycoprotein 1 (orosomucoid 1) | ORM1 | A1AG1_MOUSE | 1 | 919 | −1.2 | 3.7 10−3 | = | / |
| Alpha-1-antitrypsin 1-1 | SERPINA1 | A1AT1_MOUSE | 1 | 357 | −1.2 | 2.0 10−2 | −1.2 | 4.7 10−2 |
| Alpha-1-antitrypsin 1-2 | SERPINA1 | A1AT2_MOUSE | 1 | 683 | −1.5 | 2.0 10−3 | −1.4 | 2.4 10−2 |
| Alpha-2-HS-glycoprotein (Fetuin A) | AHSG | FETUA_MOUSE | 2 | 713, 849 | −1.2 | 8.4 10−4 | = | - |
| Alpha-2-macroglobulin | A2M | A2M_MOUSE | 3 | 996, 1009, 1010 | = | / | −1.5 | 5.4 10−4 |
| Antithrombin-III | SERPINC1 | ANT3_MOUSE | 3 | 1695, 1697, 648 | +1.2 | 1.2 10−3 | +1.1 | 5.2 10−3 |
| Apolipoprotein A-I | APOA1 | APOA1_MOUSE | 1 | 1335 | = | / | −1.3 | 6.0 10−4 |
| Apolipoprotein E | APOE | APOE_MOUSE | 3 | 1080, 1099, 1106 | −1.2 | 6.6 10−3 | = | / |
| Apolipoprotein H (Beta-2-glycoprotein I) | APOH | APOH_MOUSE | 1 | 626 | +1.2 | 4.4 10−3 | +1.2 | 8.2 10−3 |
| Clusterin | CLU | CLUS_MOUSE | 7 | 938, 1015, 1036, 1042, 1096, 1669, 1670 | +1.3 | 2.8 10−8 | +1.2 | 5.7 10−7 |
| −1.2 | 1.2 10−3 | −1.2 | 2.2 10−2 | |||||
| Coagulation factor X | F10 | FA10_MOUSE | 1 | 778 | −1.2 | 2.2 10−3 | = | / |
| Complement C1r-A subcomponent | C1R | C1RA_MOUSE | 2 | 475, 477 | +1.2 | 2.3 10−3 | +1.3 | 6.3 10−5 |
| Complement C1s-A subcomponent | C1S | CS1A_MOUSE | 1 | 476 | +1.1 | 5 10−3 | = | / |
| Complement C3 | C3 | CO3_MOUSE | 4 | 579, 595, 1205, 1677 | −1.3 | 2.9 10−3 | −1.2 | 2.3 10−3 |
| Complement C4-B | C4A | CO4B_MOUSE | 1 | 1594 | −1.3 | 1.7 10−2 | = | / |
| Complement component 7 | C7 | CO7_HUMAN | 5 | 400, 401, 407, 408, 412 | −1.3 | 7.5 10−4 | −1.4 | 2.9 10−2 |
| Complement factor H | CFH | CFAH_MOUSE | 1 | 164 | −1.1 | 4.5 10−3 | = | / |
| Complement factor I | CFI | CFAI_MOUSE | 1 | 1057 | +1.1 | 1.5 10−3 | +1.2 | 2.0 10−3 |
| Fetuin B | FETUB | FETUB_MOUSE | 2 | 673, 676 | +1.2 | 1.7 10−5 | +1.1 | 2.4 10−3 |
| Group-specific component (Vitamin D binding protein) | GC | VTDB_MOUSE | 2 | 667, 678 | +1.3 | 7.4 10−4 | +1.3 | 4.3 10−3 |
| Haptoglobin | HP | HPT_MOUSE | 2 | 931, 942 | +7.1 | 2.1 10−7 | +2.5 | 4.6 10−2 |
| Hemopexin | HPX | HEMO_MOUSE | 9 | 499, 509, 511, 512, 519, 541, 578, 1681, 1692 | −1.3 | 2.3 10−3 | +1.2 | 6.9 10−3 |
| +1.3 | 4.5 10−5 | |||||||
| Ig mu chain C region secreted form | Ighm | IGHM_MOUSE | 2 | 319, 358 | −1.3 | 7.9 10−3 | −1.1 | 2.0 10−2 |
| Inter alpha-trypsin inhibitor, heavy chain 4, isoform CRA_b | ITIH4 | A6X935_MOUSE | 1 | 345 | +1.1 | 1.0 10−2 | +1.1 | 4.4 10−2 |
| Kininogen-1 | KNG1 | KNG1_MOUSE | 1 | 569 | +1.3 | 1.2 10−4 | = | / |
| Murinoglobulin-1 | Mug1 | MUG1_MOUSE | 1 | 627 | +1.1 | 2.5 10−2 | +1.1 | 2.8 10−3 |
| Novel protein similar to odorant binding protein Ia Obp1a | Gm5938 | A2AEN9_MOUSE | 1 | 1653 | +1.5 | 1.1 10−2 | = | / |
| Odorant binding protein Ia | Obp1a | P97336_MOUSE | 1 | 1632 | +1.6 | 4.2 10-3 | = | / |
| Pantothenate kinase 4 | PANK4 | PANK4_MOUSE | 1 | 1129 | +1.9 | 2.9 10-11 | +1.6 | 3.6 10−7 |
| Peroxiredoxin-2 | PRDX2 | PRDX2_MOUSE | 1 | 1413 | = | / | −1.3 | 1.6 10−2 |
| Glycosylphosphatidylinositol specific phospholipase D1 | GPLD1 | PHLD_MOUSE | 2 | 355, 520 | −1.3 | 8.0 10-4 | −1.2 | 6.4 10−3 |
| Plasminogen | PLG | PLMN_MOUSE | 2 | 309, 415 | −1.2 | 1.1 10-3 | −1.7 | 4.2 10−2 |
| +1.6 | 7.9 10-3 | +1.6 | 2.6 10−2 | |||||
| Serine protease inhibitor A3K | Serpina3k | SPA3K_MOUSE | 12 | 54, 86, 87, 256, 257, 262, 531, 588, 599, 621, 1671, 1672 | −1.6 | 2.0 10-5 | −1.3 | 4.0 10−2 |
| Serum paraoxonase/ arylesterase 1 | PON1 | PON1_MOUSE | 1 | 803 | −1.2 | 5.0 10-3 | = | / |
| Zinc-alpha-2-glycoprotein | AZGP1 | ZA2G_MOUSE | 2 | 787, 790 | +1.1 | 3.1 10-2 | +1.1 | 4.1 10−4 |

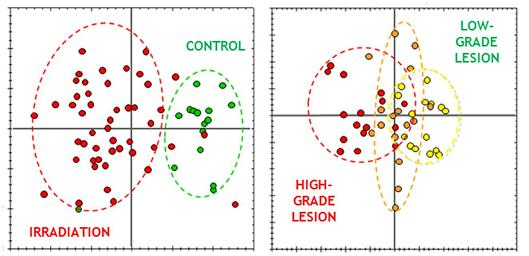
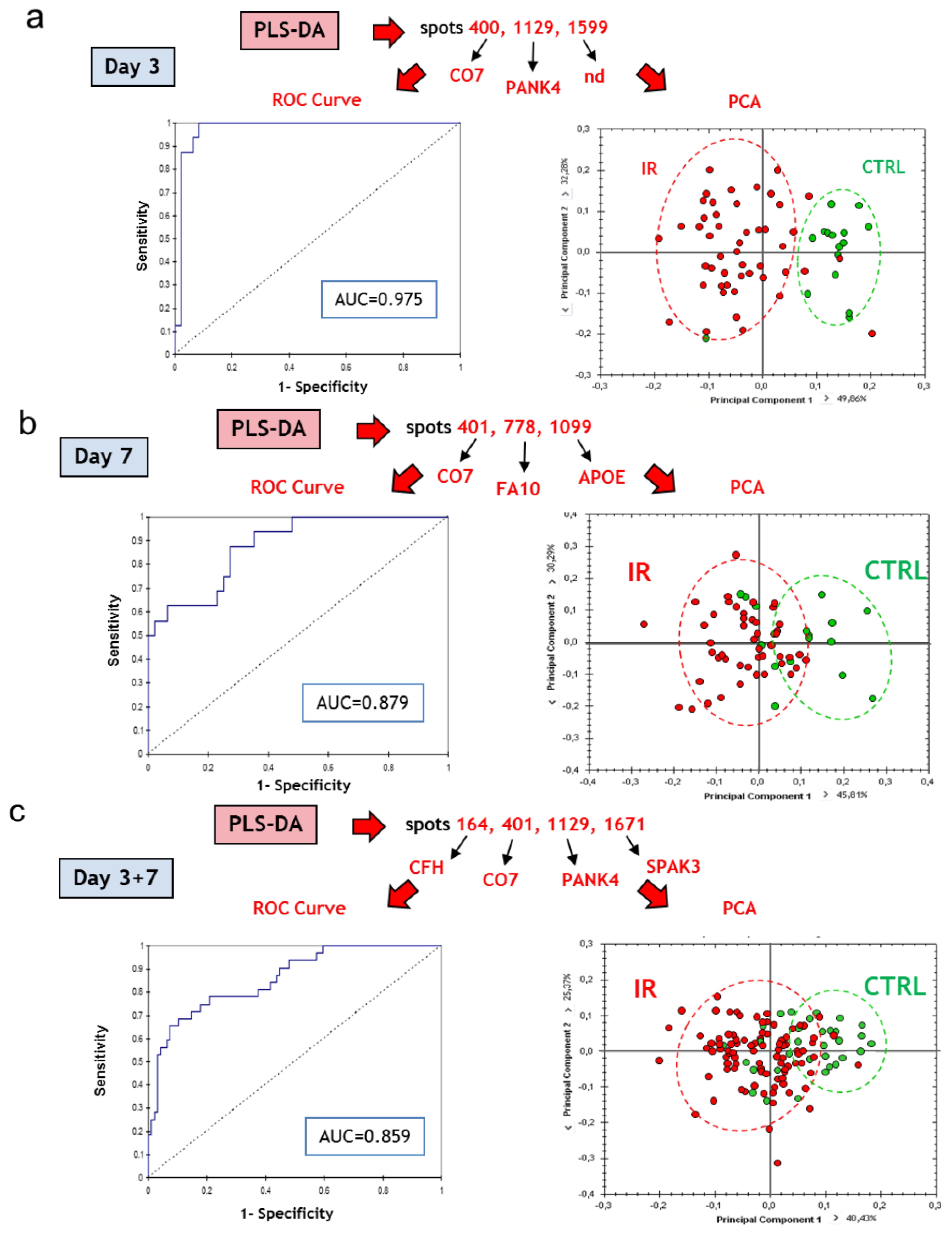
3.3.4. Multivariate Statistical Analysis of 2D-DIGE Data: Best Candidates for Triage and Prognosis
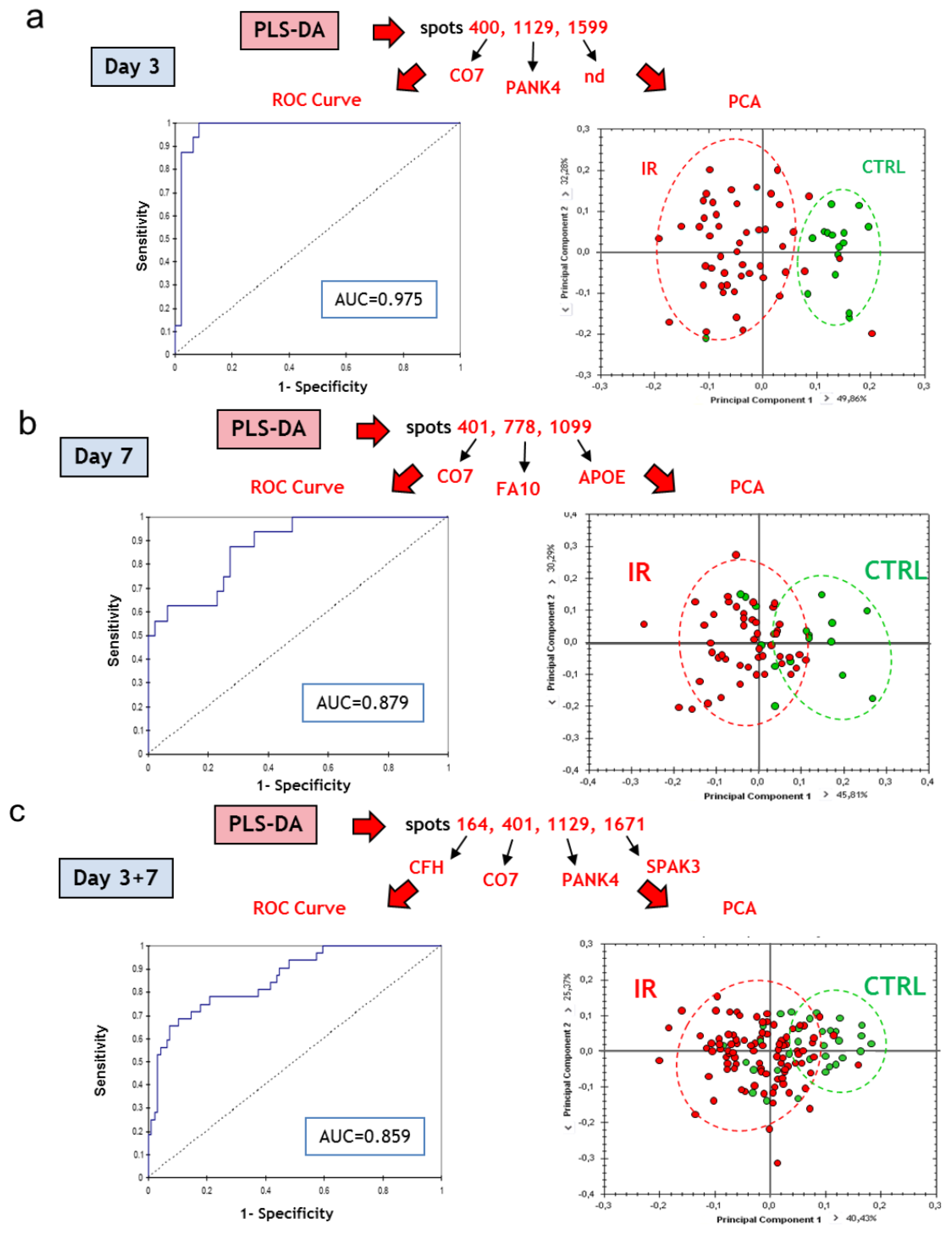
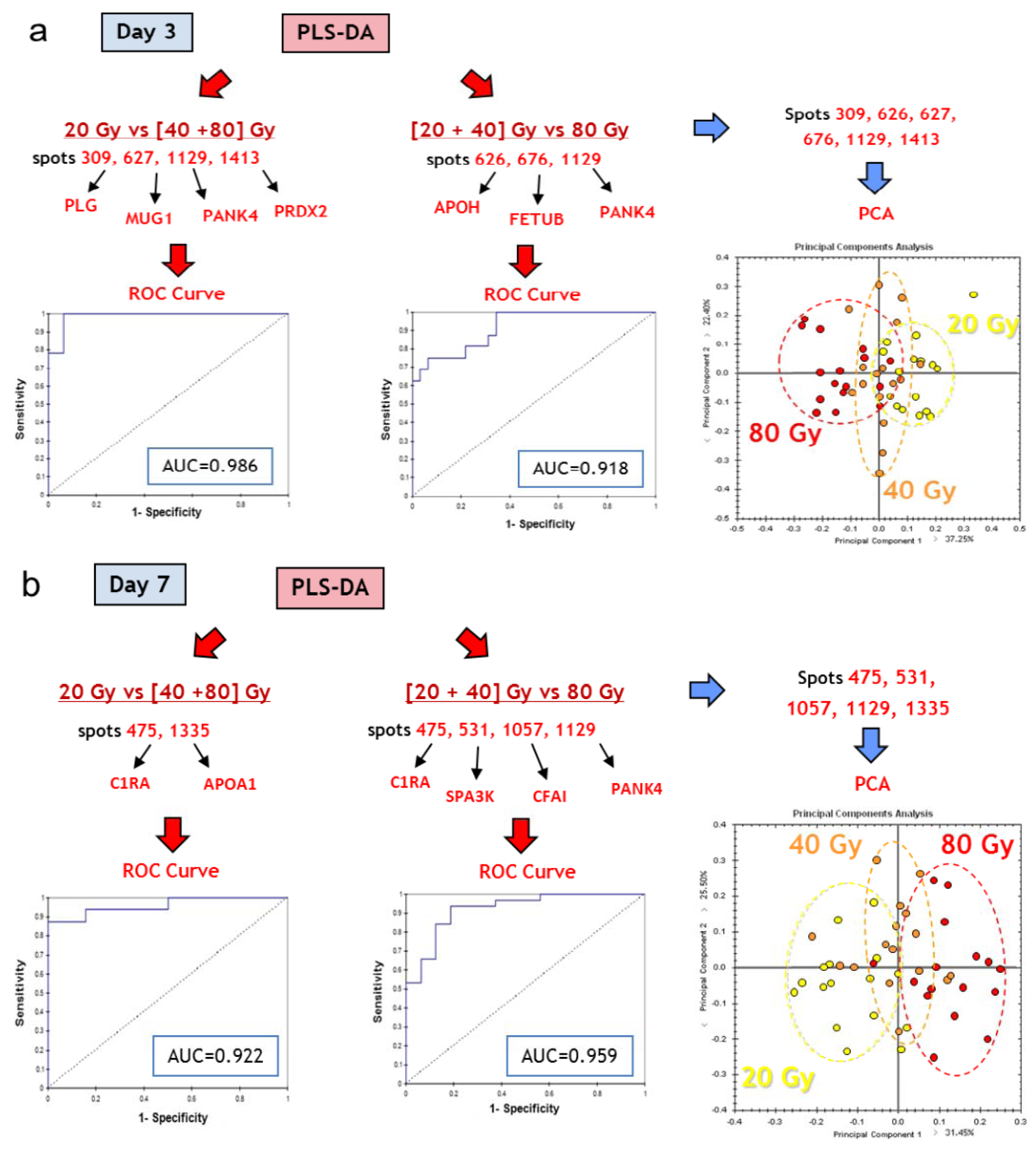
| Best protein candidates based on PLS-DA | |||||||
|---|---|---|---|---|---|---|---|
| Protein name | Accession (Uniprot KB) | Name in DB | Spot number 1 | Irradiation 2,3 | Dose 2,4 | ||
| Max fold change (p < 0.05) | p-value | Max fold change (p < 0.05) | p-value | ||||
| Apolipoprotein A-I | APOA1_MOUSE | APOA1 | 1335 | = | / | −1.3 | 6.0 10−4 |
| Apolipoprotein E | APOE_MOUSE | APOE | 1099 | −1.2 | 1.0 10−2 | = | / |
| Apolipoprotein H | APOH_MOUSE | APOH | 626 | +1.2 | 4.4 10−3 | +1.2 | 8.2 10−3 |
| Coagulation factor X | FA10_MOUSE | F10 | 778 | −1.2 | 2.2 10−3 | = | / |
| Complement C1r-A subcomponent | C1RA_MOUSE | C1R | 475 | = | / | +1.3 | 6.3 10−5 |
| Complement component 7 | CO7_HUMAN | C7 | 400, 401 | −1.3 | 2.5 10−4 | = | / |
| Complement factor H | CFAH_MOUSE | CFH | 164 | +1.1 | 4.5 10−3 | = | / |
| Complement factor I | CFAI_MOUSE | CFI | 1057 | −1.1 | 1.5 10−3 | +1.2 | 2.0 10−3 |
| Fetuin B | FETUB_MOUSE | FETUB | 676 | +1.2 | 1.7 10−5 | +1.1 | 2.4 10−3 |
| Murinoglobulin 1 | MUG1_MOUSE | Mug1 | 627 | +1.1 | 2.5 10−2 | +1.1 | 2.8 10−3 |
| Pantothenate kinase 4 | PANK4_MOUSE | PANK4 | 1129 | +1.9 | 2.9 10−11 | +1.6 | 3.6 10−7 |
| Peroxiredoxin-2 | PRDX2_MOUSE | PRDX2 | 1413 | = | - | −1.3 | 1.6 10−2 |
| Plasminogen | PLMN_MOUSE | PLG | 309 | −1.2 | 1.1 10−3 | −1.3 | 4.2 10−3 |
| Serine protease inhibitor A3K | SPA3K_MOUSE | Serpina3k | 1671 | −1.6 | 2.0 10−3 | −1.3 | 4.0 10−2 |
| Non-identified | - | - | 1599 | +1.7 | 1.8 10−3 | = | / |
| Results for ROC curve analysis | |||
|---|---|---|---|
| Discriminant variables | Group discrimination | AUC (ROC) | |
| C7/PANK4/spot 1599 | Irradiation | NI vs. IR, d3 | 0.975 |
| C7/F10/APOE | NI vs. IR, d7 | 0.879 | |
| C7/PANK4/Serpina3k/CFH | NI vs. IR, d3+d7 | 0.859 | |
| Mug1/PANK4/PRDX2/PLG | Dose | 20 Gy vs. (40+80) Gy, d3 | 0.986 |
| APOH/FETUB/PANK4 | (20+40) Gy vs. 80 Gy, d3 | 0.918 | |
| C1RA/APOA1 | 20 Gy vs. (40+80) Gy, d7 | 0.922 | |
| C1RA/Serpina3k/CFI/PANK4 | (20+40) Gy vs. 80 Gy, d7 | 0.959 | |
4. Conclusions
Supplementary Materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgments
Conflicts of Interest
References
- Hopewell, J.W. The skin: Its structure and response to ionizing radiation. Int. J. Radiat. Biol. 1990, 57, 751–773. [Google Scholar] [CrossRef]
- Gottlober, P.; Bezold, G.; Weber, L.; Gourmelon, P.; Cosset, J.M.; Bahren, W.; Hald, H.J.; Fliedner, T.M.; Peter, R.U. The radiation accident in Georgia: Clinical appearance and diagnosis of cutaneous radiation syndrome. J. Am. Acad. Dermatol. 2000, 42, 453–458. [Google Scholar] [CrossRef]
- Peter, R.U.; Steinert, M.; Gottlober, P. The cutaneous radiation syndrome: Diagnosis and treatment. Radioprotection 2001, 49, 451–457. [Google Scholar]
- Muller, K.; Meineke, V. Advances in the management of localized radiation injuries. Health Phys. 2010, 98, 843–850. [Google Scholar] [CrossRef]
- Meineke, V. The role of damage to the cutaneous system in radiation-induced multi-organ failure. Br. J. Radiol. Suppl. 2005, 27, 85–99. [Google Scholar]
- Peter, R.U. Cutaneous radiation syndrome in multi-organ failure. Br. J. Radiol. 2005, 27, 180–184. [Google Scholar] [CrossRef]
- Ainsbury, E.A.; Bakhanova, E.; Barquinero, J.F.; Brai, M.; Chumak, V.; Correcher, V.; Darroudi, F.; Fattibene, P.; Gruel, G.; Guclu, I.; et al. Review of retrospective dosimetry techniques for external ionising radiation exposures. Radiat. Prot. Dosimetry 2011, 147, 573–592. [Google Scholar] [CrossRef]
- Lataillade, J.J.; Doucet, C.; Bey, E.; Carsin, H.; Huet, C.; Clairand, I.; Bottollier-Depois, J.F.; Chapel, A.; Ernou, I.; Gourven, M.; et al. New approach to radiation burn treatment by dosimetry-guided surgery combined with autologous mesenchymal stem cell therapy. Regen. Med. 2007, 2, 785–794. [Google Scholar] [CrossRef]
- Bey, E.; Prat, M.; Duhamel, P.; Benderitter, M.; Brachet, M.; Trompier, F.; Battaglini, P.; Ernou, I.; Boutin, L.; Gourven, M.; et al. Emerging therapy for improving wound repair of severe radiation burns using local bone marrow-derived stem cell administrations. Wound Repair Regen. 2010, 18, 50–58. [Google Scholar] [CrossRef]
- Akita, S.; Akino, K.; Hirano, A.; Ohtsuru, A.; Yamashita, S. Mesenchymal stem cell therapy for cutaneous radiation syndrome. Health Phys. 2010, 98, 858–862. [Google Scholar] [CrossRef]
- Benderitter, M.; Gourmelon, P.; Bey, E.; Chapel, A.; Clairand, I.; Prat, M.; Lataillade, J.J. New emerging concepts in the medical management of local radiation injury. Health Phys. 2010, 98, 851–857. [Google Scholar] [CrossRef]
- Forcheron, F.; Agay, D.; Scherthan, H.; Riccobono, D.; Herodin, F.; Meineke, V.; Drouet, M. Autologous adipocyte derived stem cells favour healing in a minipig model of cutaneous radiation syndrome. PLoS One 2012, 7, e31694. [Google Scholar]
- Friesecke, I.; Beyrer, K.; Fliedner, T.M. How to cope with radiation accidents: The medical management. Br. J. Radiol. 2001, 74, 121–122. [Google Scholar]
- Fliedner, T.M.; Friesecke, I.; Beyrer, K. Medical Management of Radiation Accident: Manual on the Acute Radiation Syndrome; British Institute of Radiology: Oxford, UK, 2001; pp. 1–66. [Google Scholar]
- Guipaud, O.; Benderitter, M. Protein biomarkers for radiation exposure: Towards a proteomic approach as a new investigation tool. Ann. Ist. Super Sanita 2009, 45, 278–286. [Google Scholar]
- Sharma, M.; Moulder, J.E. The urine proteome as a radiation biodosimeter. Adv. Exp. Med. Biol. 2013, 990, 87–100. [Google Scholar] [CrossRef]
- Guipaud, O. Serum and plasma proteomics and its possible use as detector and predictor of radiation diseases. Adv. Exp. Med. Biol. 2013, 990, 61–86. [Google Scholar] [CrossRef]
- Guipaud, O.; Holler, V.; Buard, V.; Tarlet, G.; Royer, N.; Vinh, J.; Benderitter, M. Time-course analysis of mouse serum proteome changes following exposure of the skin to ionizing radiation. Proteomics 2007, 7, 3992–4002. [Google Scholar] [CrossRef]
- Chaze, T.; Slomianny, M.C.; Milliat, F.; Tarlet, G.; Lefebvre-Darroman, T.; Gourmelon, P.; Bey, E.; Benderitter, M.; Michalski, J.C.; Guipaud, O. Alteration of the serum N-glycome of mice locally exposed to high doses of ionizing radiation. Mol. Cell Proteomics 2013, 12, 283–301. [Google Scholar] [CrossRef]
- Petricoin, E.F.; Ardekani, A.M.; Hitt, B.A.; Levine, P.J.; Fusaro, V.A.; Steinberg, S.M.; Mills, G.B.; Simone, C.; Fishman, D.A.; Kohn, E.C.; et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002, 359, 572–577. [Google Scholar] [CrossRef]
- Petricoin, E.F.; Zoon, K.C.; Kohn, E.C.; Barrett, J.C.; Liotta, L.A. Clinical proteomics: Translating benchside promise into bedside reality. Nat. Rev. 2002, 1, 683–695. [Google Scholar]
- Fung, E.T.; Yip, T.T.; Lomas, L.; Wang, Z.; Yip, C.; Meng, X.Y.; Lin, S.; Zhang, F.; Zhang, Z.; Chan, D.W.; et al. Classification of cancer types by measuring variants of host response proteins using SELDI serum assays. Int. J. Cancer 2005, 115, 783–789. [Google Scholar] [CrossRef]
- Zhang, Z.; Bast, R.C., Jr.; Yu, Y.; Li, J.; Sokoll, L.J.; Rai, A.J.; Rosenzweig, J.M.; Cameron, B.; Wang, Y.Y.; Meng, X.Y.; et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004, 64, 5882–5890. [Google Scholar] [CrossRef]
- Wadsworth, J.T.; Somers, K.D.; Cazares, L.H.; Malik, G.; Adam, B.L.; Stack, B.C., Jr.; Wright, G.L., Jr.; Semmes, O.J. Serum protein profiles to identify head and neck cancer. Clin. Cancer Res. 2004, 10, 1625–1632. [Google Scholar] [CrossRef]
- Koopmann, J.; Zhang, Z.; White, N.; Rosenzweig, J.; Fedarko, N.; Jagannath, S.; Canto, M.I.; Yeo, C.J.; Chan, D.W.; Goggins, M. Serum diagnosis of pancreatic adenocarcinoma using surface-enhanced laser desorption and ionization mass spectrometry. Clin. Cancer. Res. 2004, 10, 860–868. [Google Scholar] [CrossRef]
- Vlahou, A.; Laronga, C.; Wilson, L.; Gregory, B.; Fournier, K.; McGaughey, D.; Perry, R.R.; Wright, G.L., Jr.; Semmes, O.J. A novel approach toward development of a rapid blood test for breast cancer. Clin. Breast Cancer 2003, 4, 203–209. [Google Scholar]
- Petricoin, E.F., 3rd; Ornstein, D.K.; Paweletz, C.P.; Ardekani, A.; Hackett, P.S.; Hitt, B.A.; Velassco, A.; Trucco, C.; Wiegand, L.; Wood, K.; et al. Serum proteomic patterns for detection of prostate cancer. J. Natl. Cancer Inst. 2002, 94, 1576–1578. [Google Scholar]
- Lin, Q.; Peng, Q.; Yao, F.; Pan, X.F.; Xiong, L.W.; Wang, Y.; Geng, J.F.; Feng, J.X.; Han, B.H.; Bao, G.L.; et al. A classification method based on principal components of SELDI spectra to diagnose of lung adenocarcinoma. PLoS One 2012, 7, e34457. [Google Scholar]
- Bonneterre, J.; Revillion, F.; Desauw, C.; Blot, E.; Kramar, A.; Fournier, C.; Hornez, L.; Peyrat, J.P. Plasma and tissue proteomic prognostic factors of response in primary breast cancer patients receiving neoadjuvant chemotherapy. Oncol. Rep. 2013, 29, 355–361. [Google Scholar]
- Menard, C.; Johann, D.; Lowenthal, M.; Muanza, T.; Sproull, M.; Ross, S.; Gulley, J.; Petricoin, E.; Coleman, C.N.; Whiteley, G.; et al. Discovering clinical biomarkers of ionizing radiation exposure with serum proteomic analysis. Cancer Res. 2006, 66, 1844–1850. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of false discovery rate under dependency. Ann. Stat. 2001, 61, 1165–1188. [Google Scholar]
- Guipaud, O.; Guillonneau, F.; Labas, V.; Praseuth, D.; Rossier, J.; Lopez, B.; Bertrand, P. An in vitro enzymatic assay coupled to proteomics analysis reveals a new DNA processing activity for Ewing sarcoma and TAF(II)68 proteins. Proteomics 2006, 6, 5962–5972. [Google Scholar] [CrossRef]
- Quintana, M.; Palicki, O.; Lucchi, G.; Ducoroy, P.; Chambon, C.; Salles, C.; Morzel, M. Inter-individual variability of protein patterns in saliva of healthy adults. J. Proteomics 2009, 72, 822–830. [Google Scholar] [CrossRef]
- Poullet, P.; Carpentier, S.; Barillot, E. myProMS, a web server for management and validation of mass spectrometry-based proteomic data. Proteomics 2007, 7, 2553–2556. [Google Scholar] [CrossRef]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway studio—The analysis and navigation of molecular networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar] [CrossRef]
- Holler, V.; Buard, V.; Gaugler, M.H.; Guipaud, O.; Baudelin, C.; Sache, A.; Perez Mdel, R.; Squiban, C.; Tamarat, R.; Milliat, F.; et al. Pravastatin limits radiation-induced vascular dysfunction in the skin. J. Invest. Dermatol. 2009, 129, 1280–1291. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell Proteomics 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Robbins, M.E.; Zhao, W. Chronic oxidative stress and radiation-induced late normal tissue injury: A review. Int. J. Radiat. Biol. 2004, 80, 251–259. [Google Scholar] [CrossRef]
- Zhao, W.; Robbins, M.E. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: Therapeutic implications. Curr. Med. Chem. 2009, 16, 130–143. [Google Scholar] [CrossRef]
- Francois, A.; Milliat, F.; Guipaud, O.; Benderitter, M. Inflammation and immunity in radiation damage to the gut mucosa. Biomed. Res. Int. 2013, 2013, e123241. [Google Scholar]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar] [CrossRef]
- Milliat, F.; Francois, A.; Tamarat, R.; Benderitter, M. Role of endothelium in radiation-induced normal tissue damages. Ann. Cardiol. Angeiol. (Paris) 2008, 57, 139–148. [Google Scholar]
- Fajardo, L.F.; Berthrong, M. Vascular lesions following radiation. Pathol. Annu. 1988, 23, 297–330. [Google Scholar]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Magic, Z.; Matic-Ivanovic, S.; Savic, J.; Poznanovic, G. Ionizing radiation-induced expression of the genes associated with the acute response to injury in the rat. Radiat. Res. 1995, 143, 187–193. [Google Scholar] [CrossRef]
- Trutic, N.; Magic, Z.; Urosevic, N.; Krtolica, K. Acute-phase protein gene expression in rat liver following whole body X-irradiation or partial hepatectomy. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2002, 133, 461–470. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Campbell, I.L.; Sun, J.R.; Withers, H.R.; McBride, W.H. Induction of acute phase gene expression by brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 619–626. [Google Scholar] [CrossRef]
- Goltry, K.L.; Epperly, M.W.; Greenberger, J.S. Induction of serum amyloid A inflammatory response genes in irradiated bone marrow cells. Radiat. Res. 1998, 149, 570–578. [Google Scholar] [CrossRef]
- Chen, C.; Lorimore, S.A.; Evans, C.A.; Whetton, A.D.; Wright, E.G. A proteomic analysis of murine bone marrow and its response to ionizing radiation. Proteomics 2005, 5, 4254–4263. [Google Scholar] [CrossRef]
- Cengiz, M.; Akbulut, S.; Atahan, I.L.; Grigsby, P.W. Acute phase response during radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 1093–1096. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H. How do tissues respond to damage at the cellular level? The role of cytokines in irradiated tissues. Radiat. Res. 1998, 150, S109–S120. [Google Scholar] [CrossRef]
- Muller, K.; Meineke, V. Radiation-induced alterations in cytokine production by skin cells. Exp. Hematol. 2007, 35, 96–104. [Google Scholar] [CrossRef]
- Benderitter, M.; Isoir, M.; Buard, V.; Durand, V.; Linard, C.; Vozenin-Brotons, M.C.; Steffanazi, J.; Carsin, H.; Gourmelon, P. Collapse of skin antioxidant status during the subacute period of cutaneous radiation syndrome: A case report. Radiat. Res. 2007, 167, 43–50. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chaze, T.; Hornez, L.; Chambon, C.; Haddad, I.; Vinh, J.; Peyrat, J.-P.; Benderitter, M.; Guipaud, O. Serum Proteome Analysis for Profiling Predictive Protein Markers Associated with the Severity of Skin Lesions Induced by Ionizing Radiation. Proteomes 2013, 1, 40-69. https://doi.org/10.3390/proteomes1020040
Chaze T, Hornez L, Chambon C, Haddad I, Vinh J, Peyrat J-P, Benderitter M, Guipaud O. Serum Proteome Analysis for Profiling Predictive Protein Markers Associated with the Severity of Skin Lesions Induced by Ionizing Radiation. Proteomes. 2013; 1(2):40-69. https://doi.org/10.3390/proteomes1020040
Chicago/Turabian StyleChaze, Thibault, Louis Hornez, Christophe Chambon, Iman Haddad, Joelle Vinh, Jean-Philippe Peyrat, Marc Benderitter, and Olivier Guipaud. 2013. "Serum Proteome Analysis for Profiling Predictive Protein Markers Associated with the Severity of Skin Lesions Induced by Ionizing Radiation" Proteomes 1, no. 2: 40-69. https://doi.org/10.3390/proteomes1020040
APA StyleChaze, T., Hornez, L., Chambon, C., Haddad, I., Vinh, J., Peyrat, J.-P., Benderitter, M., & Guipaud, O. (2013). Serum Proteome Analysis for Profiling Predictive Protein Markers Associated with the Severity of Skin Lesions Induced by Ionizing Radiation. Proteomes, 1(2), 40-69. https://doi.org/10.3390/proteomes1020040