1. Introduction
With the rise of faster and cheaper computers over the past few decades, the ability of scientists to use computers as a means to supplement physical experiments has burgeoned. In the world of molecular modeling, this growth has meant the emergence of many different types of software that better represent molecules with reduced difficulty for the researcher [
1]. Many tasks that used to require large, multimillion-dollar computational resources can now be performed on personal computers. In the realm of molecular recognition, this is no less true, and many docking and molecular recognition-oriented programs have been produced with great attention from the research community. As seen in
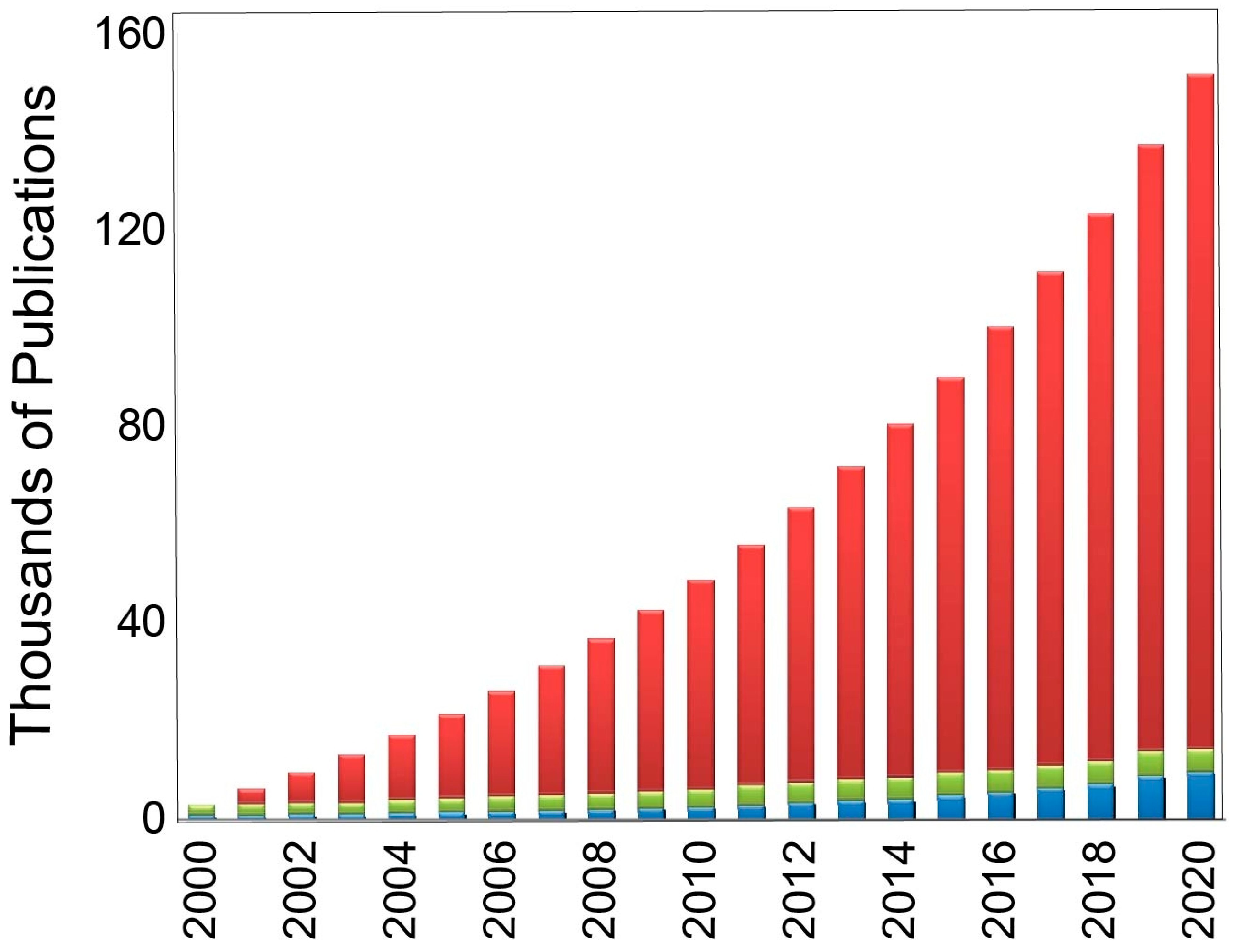
Figure 1, the number of journal articles that include the terms “docking” and/or “molecular recognition” has increased steadily and exponentially since the year 2000, with the success and use of such programs no doubt contributing.
Indeed, molecular recognition is a concept that that has garnered great interest in many scientific arenas outside of pure chemistry, including the fields of molecular physics and biophysics [
2], structural biology [
3,
4], and biochemistry [
5,
6]. While this phenomenon plays an important role in many biological systems and processes, such as receptor–ligand, antigen–antibody, and protein–nucleic acid interactions, the degree to which applications of this nature remain untreated in undergraduate curricula is noteworthy. This is particularly the case given that molecular docking is a principal component in computer-aided drug design (CADD), which we speculate is universally employed within the 1462 billion USD/year pharmaceutical industry [
7,
8,
9]. With the development of a single drug taking between 10 and 15 years and costing an average of USD 800 million, CADD is ubiquitous in reducing both the cost and time of bringing a drug to market [
10,
11].
Unfortunately, the vast majority of undergraduate STEM majors will enter graduate school or the workforce without having ever been introduced to the basic concepts of CADD and will therefore lack experience in techniques such as molecular docking. The annual mean wage for biochemists within the pharmaceutical and drug manufacturing sector in the United States was USD 98,490 between 2019 and 2021, more than triple the 2019 median income for a United States citizen [
12,
13]. For many students, especially those coming from less affluent socioeconomic backgrounds, building experience in valuable career tools may enable a greater degree of upward mobility. We thus aim herein to address this gulf by presenting a hands-on computational laboratory activity or at-home exercise that employs widely available freeware to illustrate the fundamental ideas behind molecular recognition via the popular docking software AutoDock Version 4.2.6 [
14]. Moreover, our easily adopted and easily modified activity integrates multiple disciplines in the physical, chemical, and biological sciences and can be easily adapted to serve a specific audience in any of these areas while being designed for integration into interdisciplinary curricula at the undergraduate and/or graduate level.
Though the concepts behind molecular recognition and docking can be abstract, positive outcomes from the integration of molecular modeling laboratories into existing undergraduate science curricula have been reported by both Hayes [
15] and Springer [
16]. In the study by Hayes et al., student feedback was considerably positive, with 90.3% (
n = 31) admitting that familiarity with the protein structure databank and 3D model manipulation teaching gave them new knowledge and skills. Springer et al. found that organic chemistry classes introducing a similar activity to Hayes outperformed the control group class. They speculated that the manipulation of 3D structures may ease the cognitive load on students who might be otherwise overstimulated converting 2D organic structures to 3D. It stands to reason that more complex systems, such as proteins bound to active site ligands, may burden students with an increased cognitive load compared to smaller organic molecules. Therefore, a modular activity that allows students to interact with and manipulate liganded protein structures could provide similar benefits to educational outcomes in understanding the microscopic details that govern molecular recognition and drug design.
There are many resources available for conducting such a project focused on molecular recognition. Some of the more popular resources include AutoDock [
14], DOCK 6 [
17,
18], and SwissDock [
19,
20]. While all of these can be very useful programs, we have chosen AutoDock Version 4.2.6, a molecular modeling program developed by the Scripps Research Institute based in La Jolla, California [
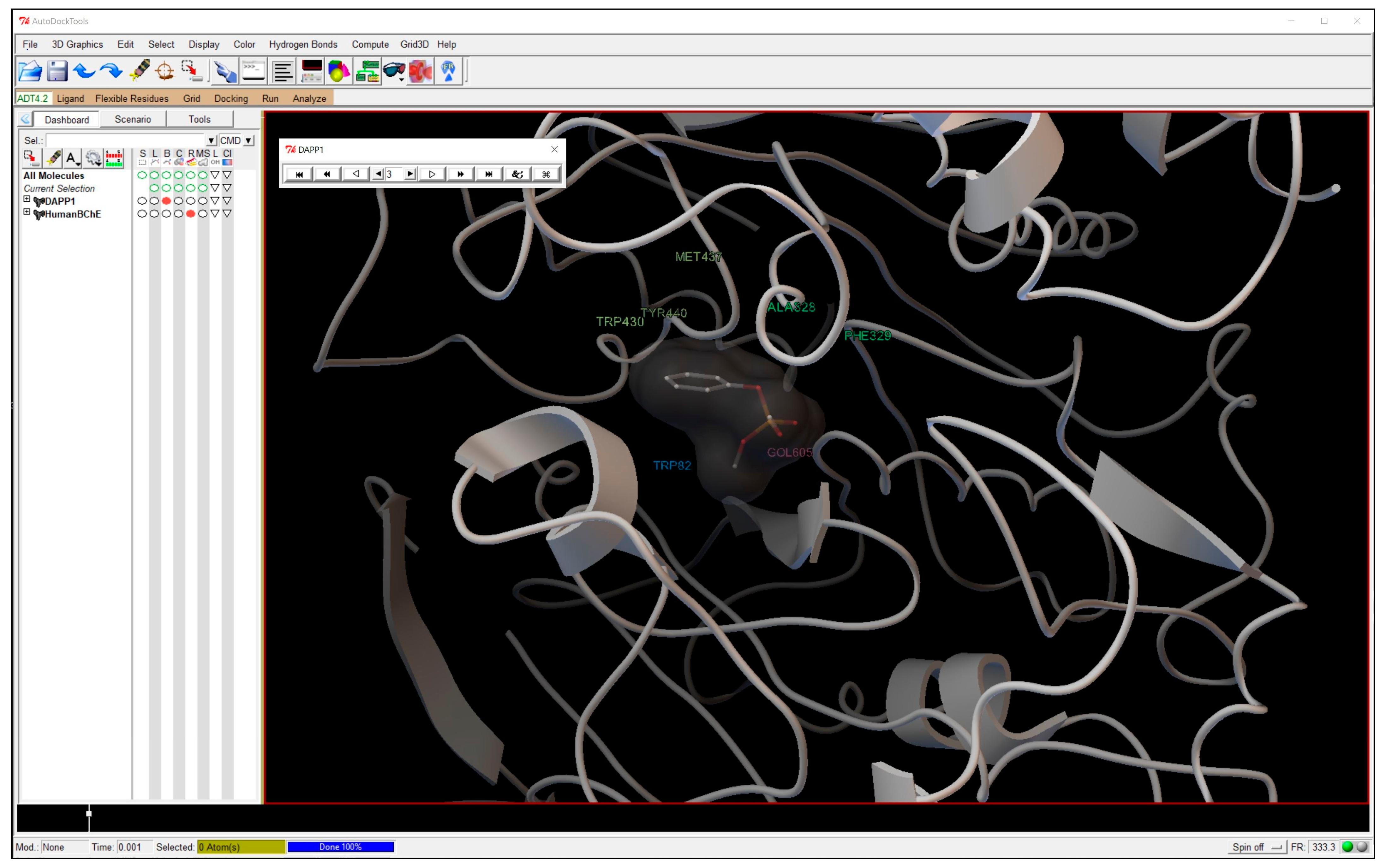
21]. According to Scripps Research Institute, AutoDock is “designed to predict how small molecules such as substrates or drug candidates, bind to a receptor of known 3D structure”. There are numerous reasons for why we favor AutoDock Version 4.2.6 over alternative modeling/docking software. First and foremost, AutoDock is distributed, with full source code, free of charge. The software has an extensive, user-friendly graphical user interface (GUI, shown in
Figure 2) and also provides a Unix-based command line interface (CLI) for those who are comfortable with command line usage. The AutoDock program provides a great platform for students to explore intermolecular interactions, giving them the opportunity to view molecules in three dimensions and practice low-complexity docking. In addition, for those encountering difficulties in using the program, the Scripps Research Institute has created a very detailed AutoDock user manual accessible at their website (autodock.scripps.edu). Finally, AutoDock provides predicted binding energies, rather than abstract docking “scores” that are less intuitive, and is available for nearly all current operating systems, making this choice the most sensible for, and supportive of, independent work on the part of students and instructors alike.
Similar educational modules using prior versions of AutoDock have been previously proposed as early as 2010 [
22,
23,
24]. However, earlier modules suffer from lack of software availability on all platforms (Mac/Windows/Linux) or may only involve the evaluation of a single drug compound, leaving no room for students to involve themselves in the drug design part of CADD [
22]. More modern and in-depth modules were designed for senior undergraduates and graduate students specializing in drug design [
23], which limits the accessibility to early undergraduate education and to disciplines outside of drug design. Our goal herein was to reach a wide range of disciplines, from physics and biochemistry to pharmaceuticals and toxicology, and to provide a module that could be incorporated within one to two laboratory sessions rather than redesign an entire lab class centered on this skill.
As detailed below, students who pursue this computational laboratory activity will learn to use the free MarvinSketch Version 23.17 utility [
25] to build drug molecules (enzyme inhibitors), which they will then import into the AutoDock software following preparation of their protein (which we have prepared and provided along with our exercise handout). They will then learn how to perform protein–ligand docking, which will generate a log file of the most favorable binding energies sampled. From their resulting structures, students will export the energetically most favorable docked “pose” into the ubiquitous Protein Data Bank (.pdb) format and visualize this optimum pose using AutoDock or any visualization program with which they are familiar (ICM-Browser, Jmol, PyMOL, VMD, etc.). In pursuing this activity, students will be introduced to a common general input/output scheme used in molecular modeling, gain a basic understanding of the docking process, and be exposed to more complex concepts regarding molecular recognition and drug design, as determined by the instructor. For example, while instructors in physical or computational areas may focus on molecular mechanical force fields and/or the Monte Carlo algorithm upon which docking calculations hinge, those in the biosciences may instead opt to emphasize the structure–function relationship or make connections to the various models of protein inhibition.
2. Pedagogical Goals
2.1. Course Approach
The current curriculum for most college and university level chemistry lab courses allows for two 3 h sessions each week. Completion of this module would be feasible within one to two such lab sessions or one 3 h lab session with assigned at-home work. Within the first lab session, the instructor should assist students with installing the following free software: AutoDock [
14], ICM-Browser [
26], and MarvinSketch [
25]. As not all students will have computer experience or have taken computer science courses, this may take over an hour for students to have installed this software and begin our tutorial. The instructor would also emphasize the synergy between the roles of biochemists, organic chemists, and computational chemists in creating novel drug inhibitors detailed in the module and ensure that the whole of the students understand the module’s questions related to basic inhibitor design principles.
By the end of the 3 h session, students will have generated one of the module’s three assigned inhibitors using MarvinSketch (module part A), opened their first inhibitor with a receptor in AutoDock (module part B), and identified the binding site (module part C) using the module’s instructions (and aid from the instructor as necessary). Familiarity and practice with the MarvinSketch and AutoDock software up to module part C is sufficient for students to independently complete module parts D, E, and F for all three assigned inhibitors in the module and the student’s custom designed inhibitor(s) in the event of a take-home assignment after one 3 h class session. For advanced course offerings, the above may be assigned as fully at-home work with instructor support provided via in-person or online teaching modalities. Our module is designed for in-person or fully virtual participation; therefore, the instructor could effectively provide support to students via various remote learning tools, such as screen sharing and a virtual white board, among others.
Upon completion of the entire module, students will have repeated the following process a minimum of four times (three module inhibitors plus one inhibitor designed by the students), as labeled according to each section in our module:
- A.
Creating a three-dimensional inhibitor in MarvinSketch;
- B.
Opening the inhibitor with a receptor protein in AutoDock;
- C.
Identifying the active site;
- D.
Performing docking calculations and generating binding energies to compare inhibitors;
- E.
Viewing ligand conformations in the active site; and
- F.
Exporting docked structures to view in external visualization software (ICM-Browser used herein).
This repetition (see
Figure 3 below) is intended to enhance retention of this process and allow the students enough attempts to achieve the module’s learning outcomes beyond following a script. Students will receive immediate feedback on how their designed drugs compared to the sampled three provided for them, challenging and improving their mental model of the drug design process [
27]. Upon completion of the lab activity, the instructor may invite students to share the docking results of their custom designed inhibitors with their classmates. With a multitude of inhibitor comparisons, students will be able to see which inhibitors outperformed the three given in the module and speculate which functional groups may be contributing to better docking scores. Sharing results and discussing which inhibitor design choices were more or less favorable can then be carried out in a follow-up class session or as an at-home group writeup.
2.2. Pedagogical Aims and Learning Outcomes
This module is intended for students who have completed general or AP chemistry curricula, the first term of organic chemistry, and an introductory molecular biology course at a minimum. Fundamental prerequisite concepts from general chemistry include types of covalent and non-covalent bonding, such as hydrogen bonding and London dispersion, and basic thermodynamic quantities, including entropy and free energy as driving forces for spontaneity. Analogous fundamental concepts from organic chemistry include basic molecular structure, three-dimensional manipulation of small compounds, and understanding how different chemical groups might play a role in binding to specific active site residues. While this module reinforces these concepts for students who lack this minimum understanding, computer-aided drug design and, by extension, the computational docking performed in this module, are inherently multidisciplinary. Students without the desired minimal knowledge may fail to appreciate the following student learning outcomes:
Understand that drug design is similar to a relay race where scientists of different disciplines hand their work to the next group in the pipeline [
28];
Gain hands-on exposure to software relevant to that CADD pipeline;
Compare quantitative docking free energies between inhibitors to determine which would make the best candidate(s) for further testing in the CADD pipeline;
Assess protein–drug interactions qualitatively to better understand relative binding strengths; and
Predict chemical modifications that would enhance the binding of potential drug species to the protein active site.
This module may be freely incorporated into lecture or lab classes in any number of biological, chemical, or physical disciplines as a computational laboratory experiment, an at-home exercise or course project, or a hybrid version of these options. Specific core concepts in related disciplines that could be taught in parallel with our module include, but are not limited to, those listed in
Figure 4. For each of these disciplines, our module invites instructors to emphasize or add additional learning objectives tailored to their course(s). For example, instructors of biochemistry-oriented classes may want to introduce point mutations to receptors outside of this module and test relevant inhibitors, while those instructing physical-chemistry-oriented coursework may emphasize binding equilibria using by-hand calculations based on predicted binding free energies.
Our module is also appropriate for integrative courses and graduate level students who may not have yet had an introduction to computational science. In addition to the binding energies resulting from docking experiments within the module, students may choose a liganded receptor for future study by molecular dynamics. While this module does not cover molecular dynamics, the authors would recommend the GROMACS molecular dynamics simulation software package [
29] available for free on Linux platforms. Other possible add-ons to this project include point-mutation-based docking, flexible receptor docking, protein–protein docking, chemical mechanism studies, and student- or group-specific studies of known inhibitor–receptor systems of interest. For example, a well-studied inhibitor–receptor system category is the interactions of toxic compounds with their respective target receptors. This approach serves as one of many effective ways to introduce students to the field of toxicology. Indeed, due to the breadth and scope of possible applications and fields discussed above, no one-size-fits-all assessment/research instrument would be appropriate with our highly interdisciplinary module.
3. Activity Overview
Students will be led step by step through the procedures following the tutorial document provided. The provided .pdb model of human butyrylcholinesterase (BChE) (see
Supplementary Materials) was prepared by removing all water, ions, and ligands from the crystal structure (PDB ID: 1P0I). Missing atoms and sidechains (none of which were near or part of the enzyme active site gorge) were added and geometry optimization on these regions was performed using BIOVIA Discovery Studio [
30]. The resulting structure was then energy-minimized, including sidechain rotomer relaxation, using the SwissPDB software [
31], as reported previously [
32,
33,
34].
While a set of test inhibitors is provided to serve as both student training and a quality control on student work, students are expected to construct one or more comparable inhibitor molecules using the MarvinSketch Version 23.17 software [
25]. After the ligand molecules are converted to 3D and read by AutoDockTools Version 1.5.7, students will load the provided BChE .pdb, which will then be converted to an extended PDB format called a PDBQT coordinate file. Once the ligand and enzyme are loaded into AutoDockTools Version 1.5.7, students will be guided through a series of steps to initiate docking calculations.
Figure 3 summarizes this process.
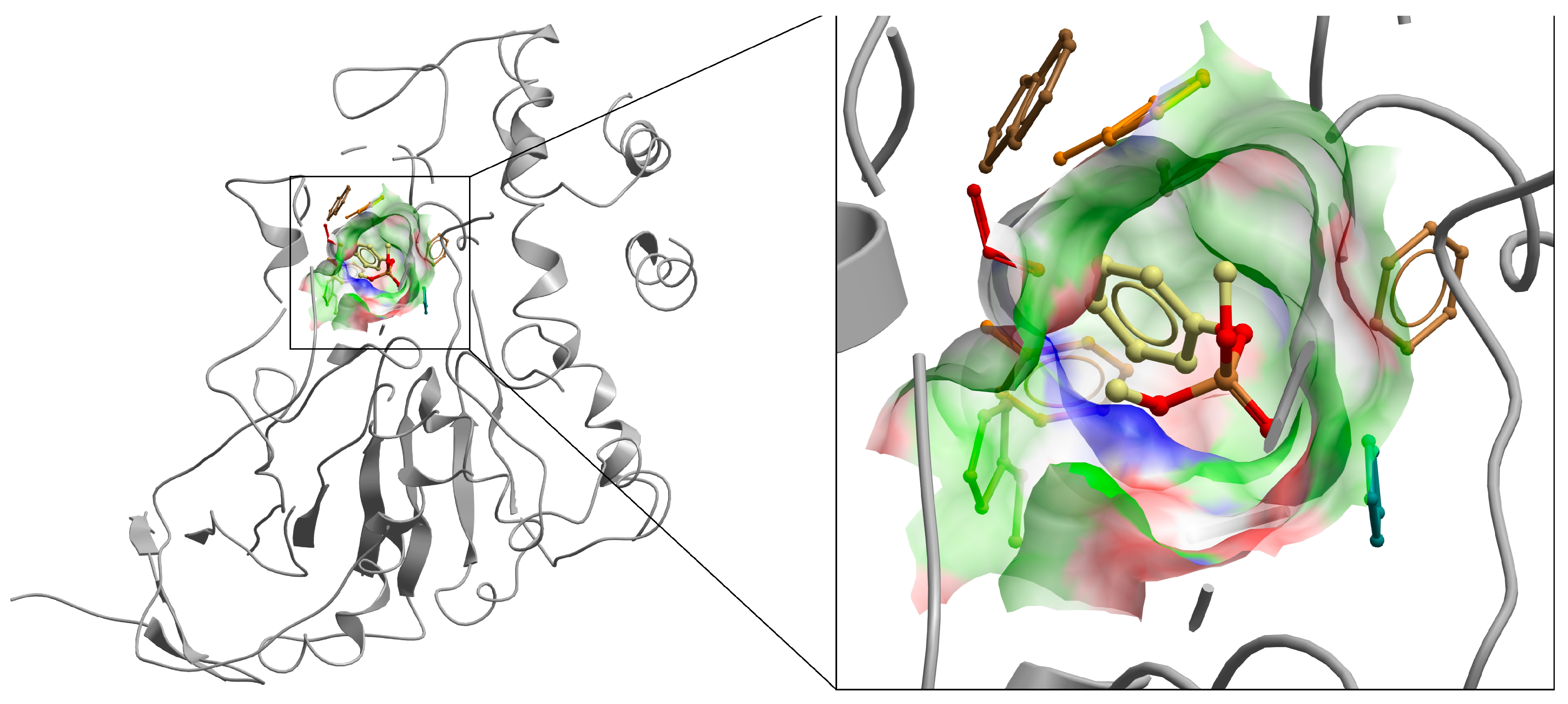
Potential binding sites of BChE will be identified using the AutoGrid module within AutoDock Version 4.2.6, which performs rapid energy evaluations of various ligand configurations within sections of the protein-termed grid points. Project files pertaining to the site will then be made by the program. For each inhibitor molecule, students will then have to set parameters for docking calculations. Once the settings are confirmed, these calculations will proceed. This should be performed at least a handful of times for each of the inhibitors to verify the consistent and accurate following of our module handout. Following docking calculations, AutoDock will generate a table of binding energies called the RMSD table that is found in the resulting DLG file at the end of the docking procedure. The RMSD table lists the binding energies of successfully docked conformations from the most to least energetically favorable. For the purposes of this project, it is not necessary to save or print this table as long as students record their lowest (most negative) docking energy. The more negative the docking energy, the better the fit and stronger the binding of the inhibitor.
Once all docking calculations are completed, students will save the docked conformation of each inhibitor with the best AutoDock docking energy. Using the AutoDockTools Version 1.5.7 window (or with another visualization software, if desired) students will then examine the 3D structure of the docked complex to look for potential interactions between each inhibitor molecule and the BChE active site. Once a student is confident in their observations, they should compare the positioning of multiple inhibitors to explain why there is or is not a significant difference in predicted AutoDock binding energies. Following complete analysis of all docked inhibitors, students will then be ready to answer follow-up questions in the course project document.
5. Conclusions
We present herein a readily adaptable method for designing novel inhibitors of the BChE enzyme and for evaluating the effective binding affinity of known BChE inhibitors by employing molecular docking using the AutoDock software. Our module, which can be easily replicated, may be incorporated into teaching laboratories across a wide range of scientific disciplines. Once this molecular docking module is completed, participating students will have attained a grasp of the fundamental aspects of computer-aided drug design. This module was designed to accomplish multiple goals. First and foremost, we aimed to introduce students to various freely available software that can be utilized to model the inhibition of BChE, or the binding to other proteins, by organic drug molecules and to generate figures that meet publication standards. Additionally, this module seeks to provide students with the essential knowledge of how different functional groups of organic drug molecules can impact binding affinity through their interactions with key residues within the active site of the enzyme through direct application of concepts taught in organic chemistry.
This module is particularly well suited to familiarize students with the important but often under-represented study of computational methods, specifically docking, in undergraduate education within the chemical, biochemical, and biophysical sciences. Consequently, we envision our module as bridging the gap between physical and/or biological disciplines at the undergraduate level and the ever-growing field of computational science. The computational method presented in this module can be integrated into both introductory science courses, such as general chemistry and biology, as well as more advanced courses, such as physical chemistry or biophysics, to allow students to participate in a hands-on activity and explore the principles of molecular recognition with minimal modifications. For instance, our module offers students an opportunity to visually explore the optimal geometries of simple organic molecules that bind within the BChE active site. This goes beyond the pen and paper approach usually covered in introductory science courses, such as Lewis dot structures and the fundamentals of intermolecular interactions, by providing students with a 3D visualization of the resulting conformation and the ability to experiment with drug molecules of their own design.
Additionally, this exercise can be used to facilitate and/or reinforce student understanding of more advanced essential concepts such as the relationship between protein structure and function, functional group chemistry and non-covalent interactions, and the thermodynamics of binding. Such concepts may be presented in introductory science coursework and become particularly crucial to success in advanced courses, such as physical chemistry, biochemistry, and biophysical course offerings. Finally, it is worth emphasizing that the computational modeling of ligand–enzyme interactions can provide valuable insights into drug binding mechanisms when experimental data are limited or not available. As such, introducing students to the applications of computational drug design can have significant benefits for their future studies and careers.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}