
Lifetime of Catalyst under Voltage Cycling in Polymer Electrolyte Fuel Cell Due to Platinum Oxidation and Dissolution



Abstract
:1. Introduction
2. Materials and Methods
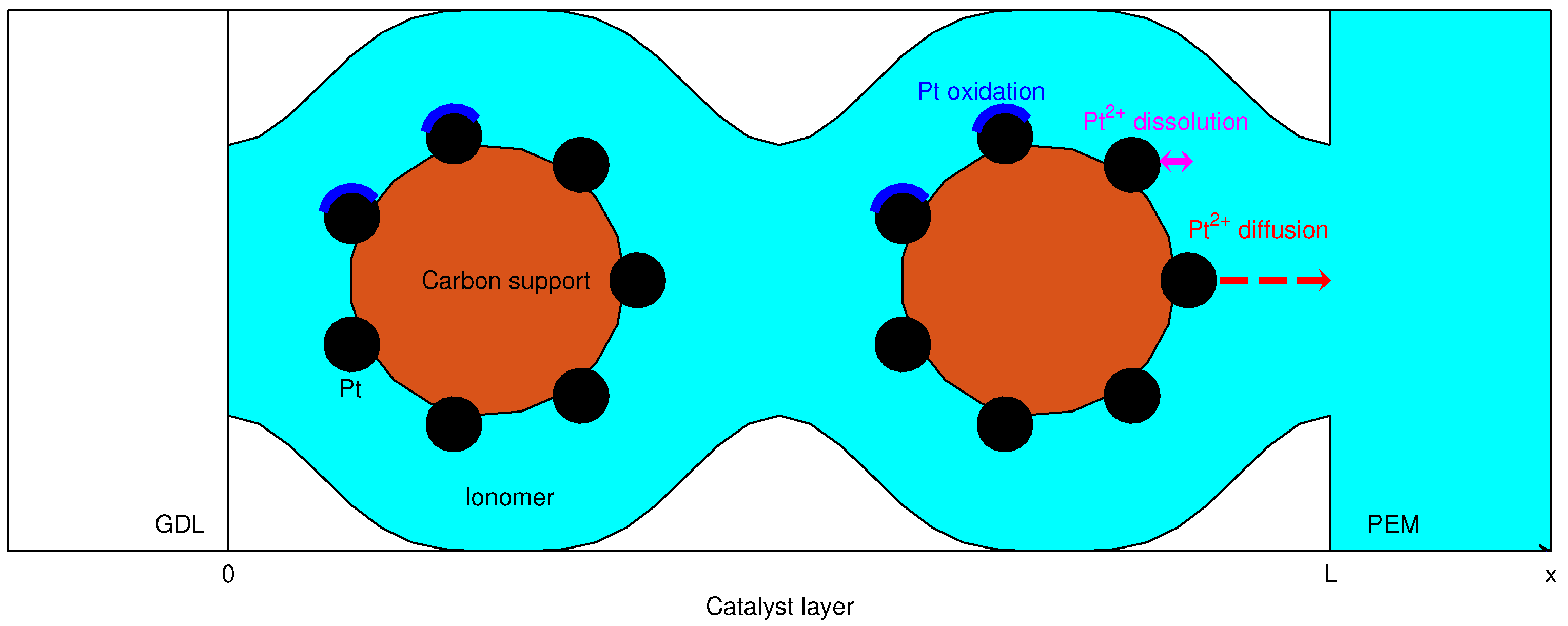
2.1. Governing Relations
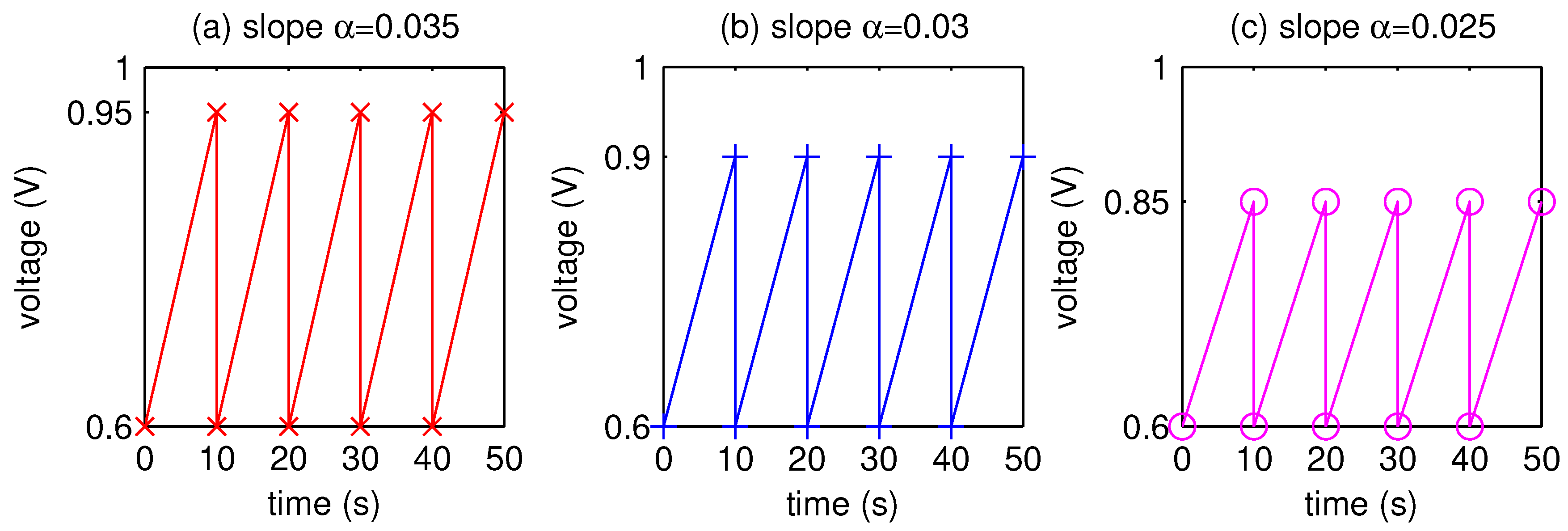
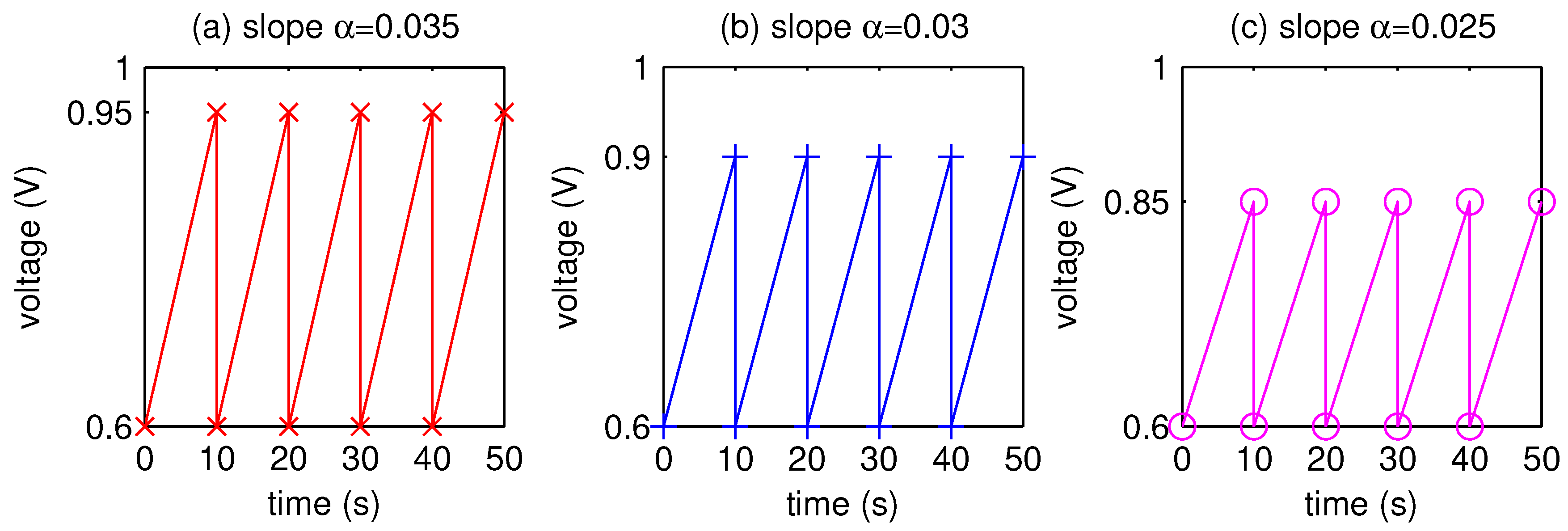
2.2. Numerical Simulation
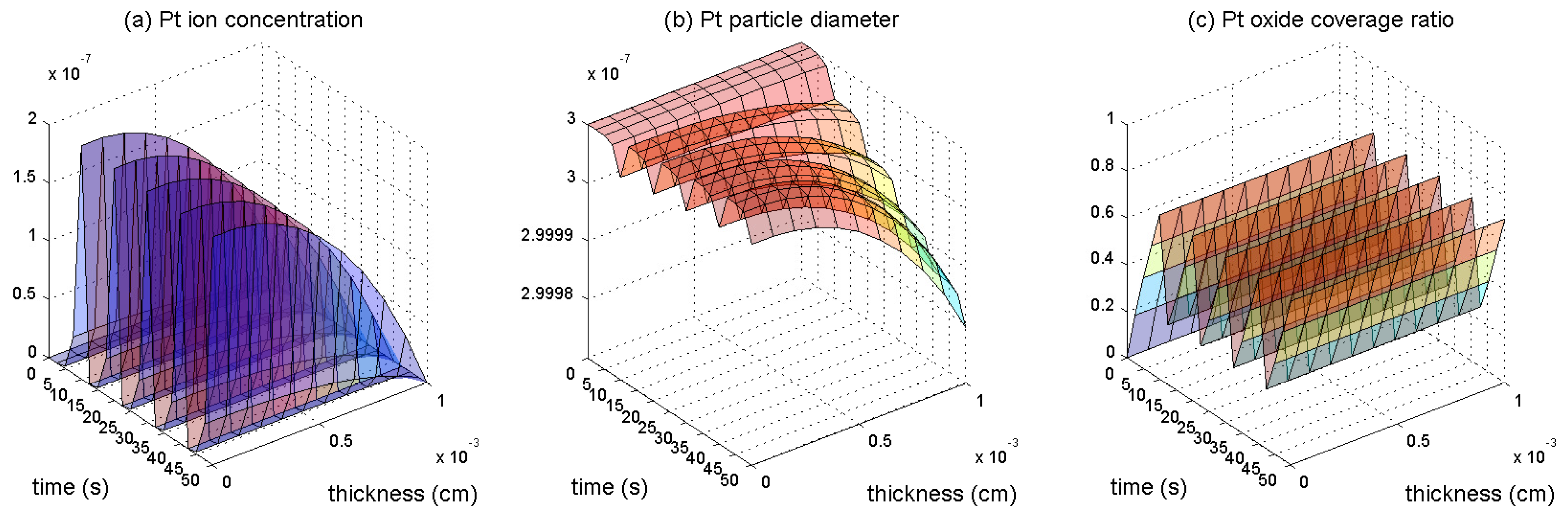
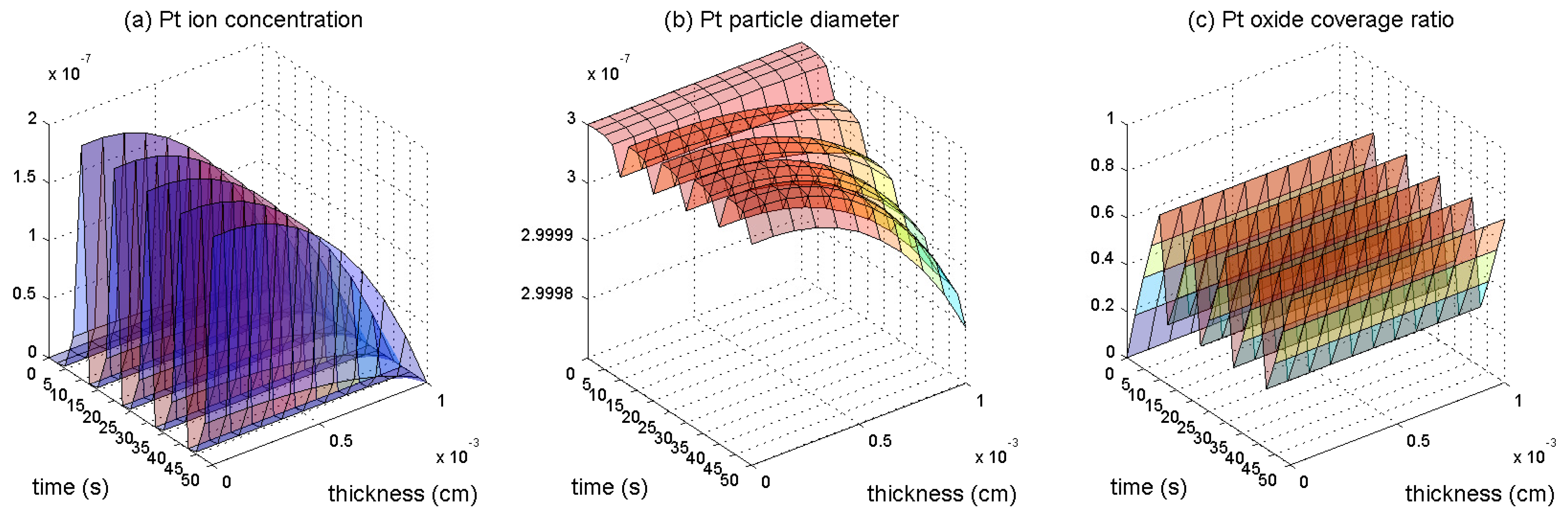
3. Results and Discussion
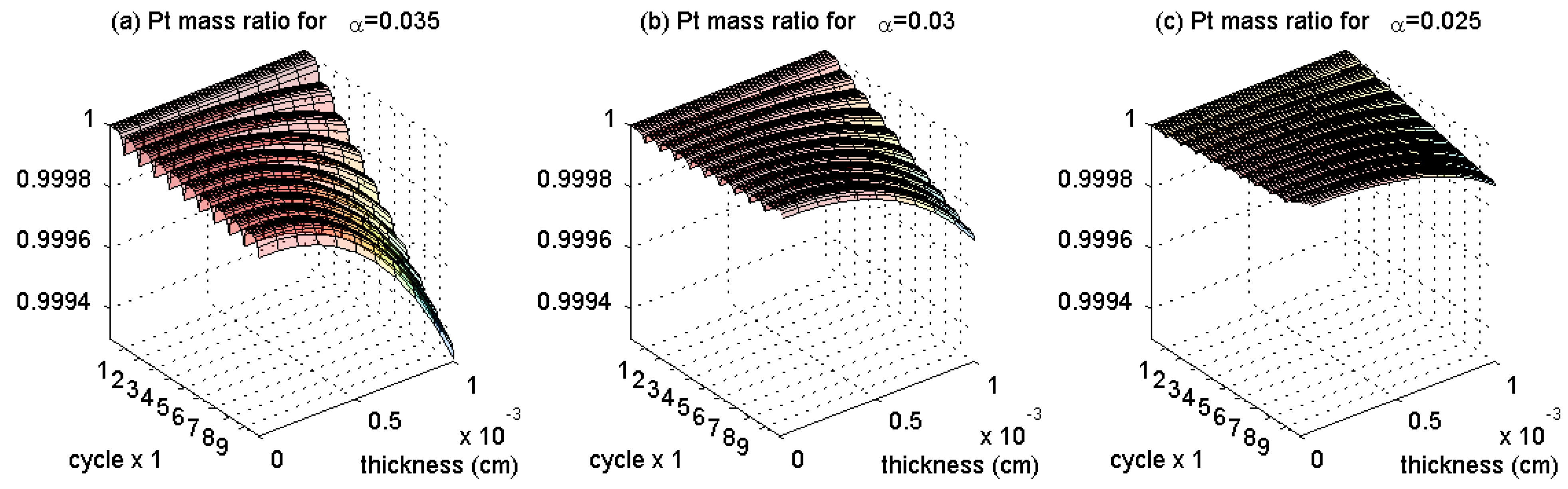
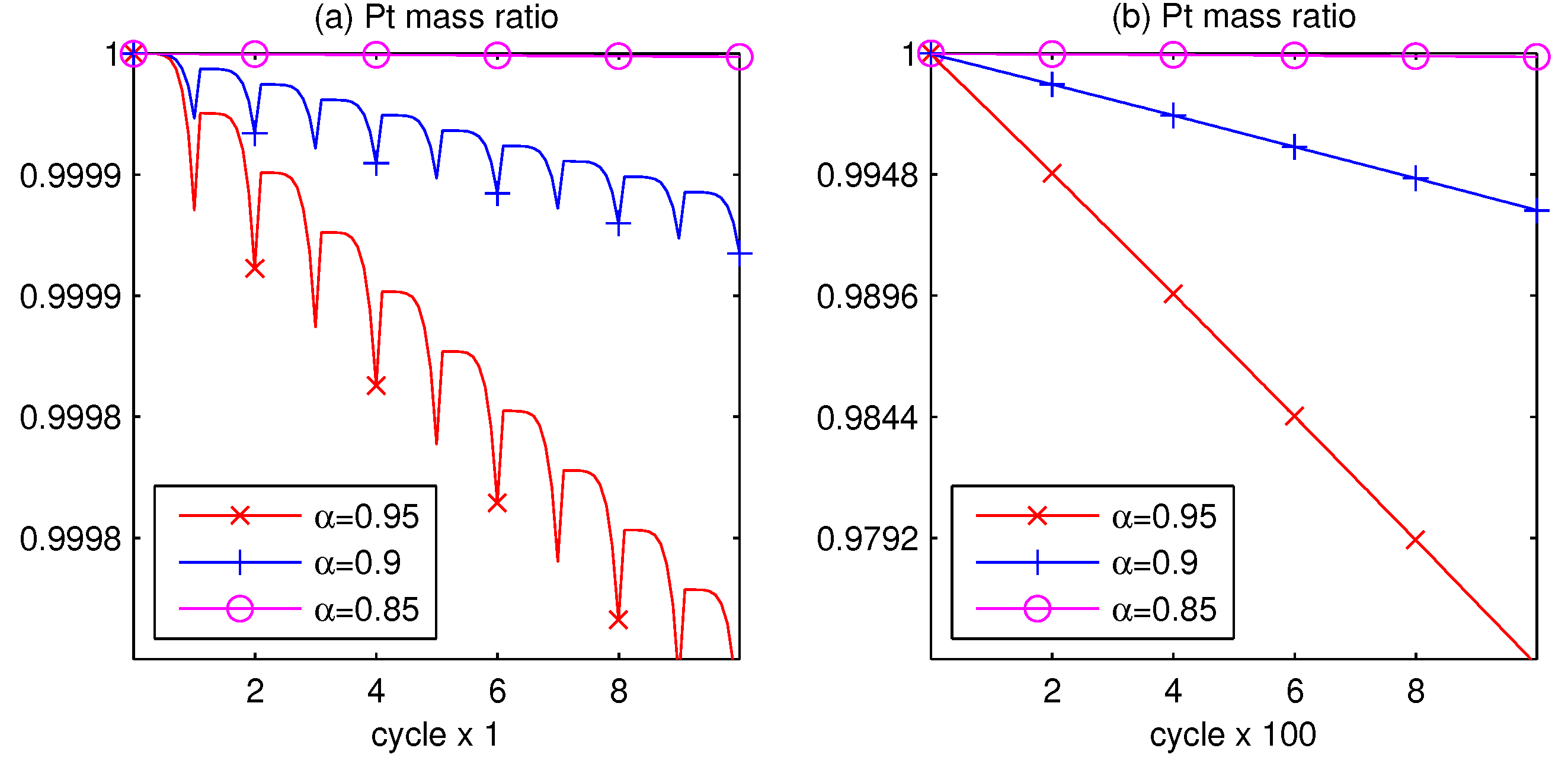
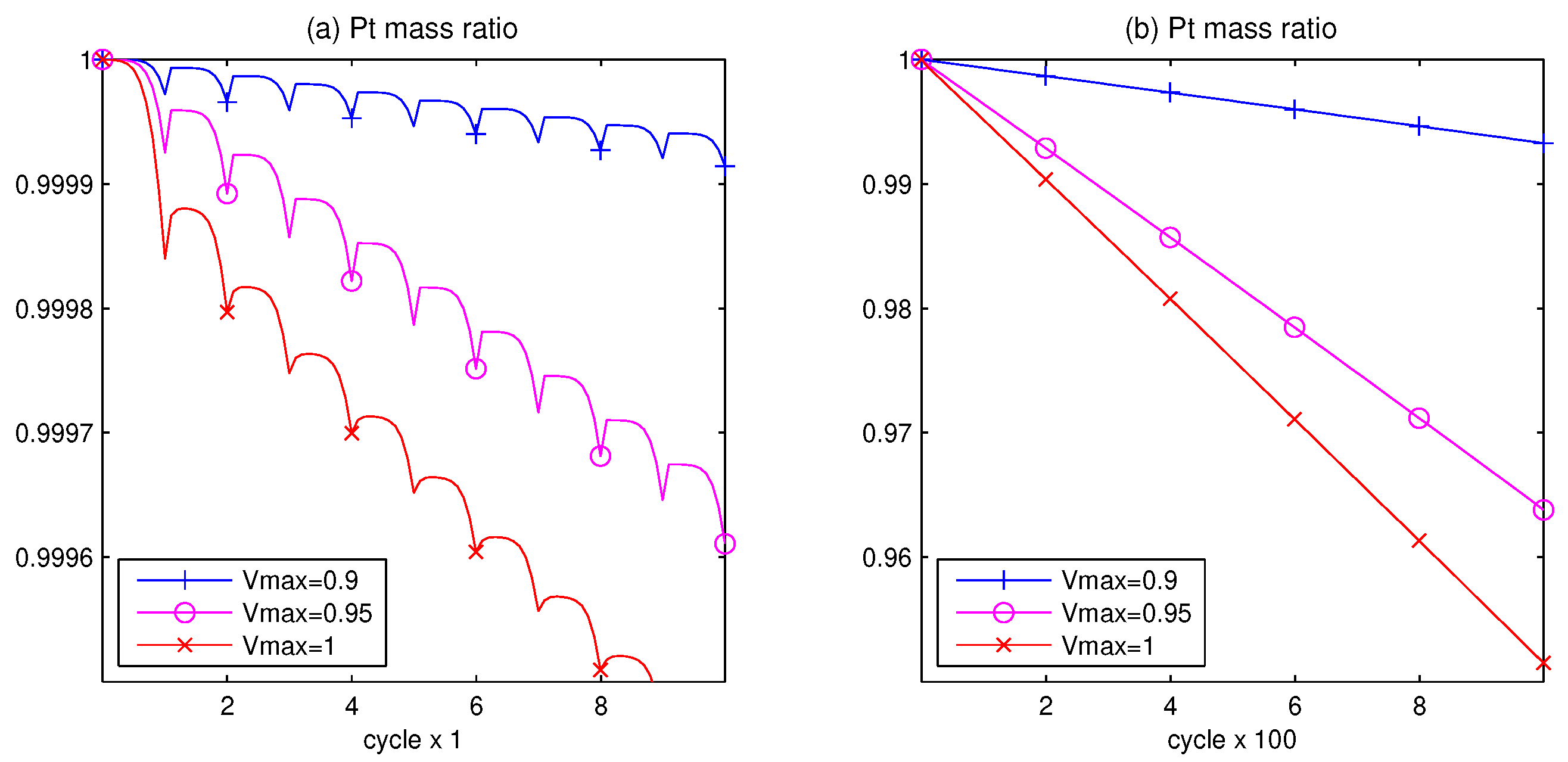
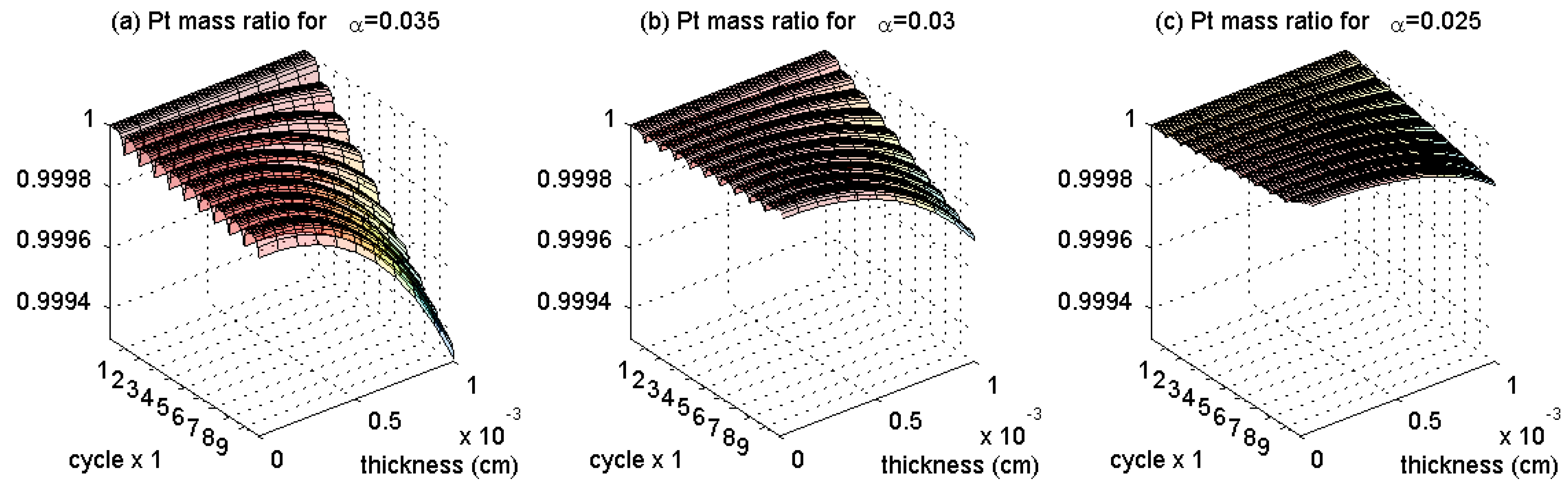
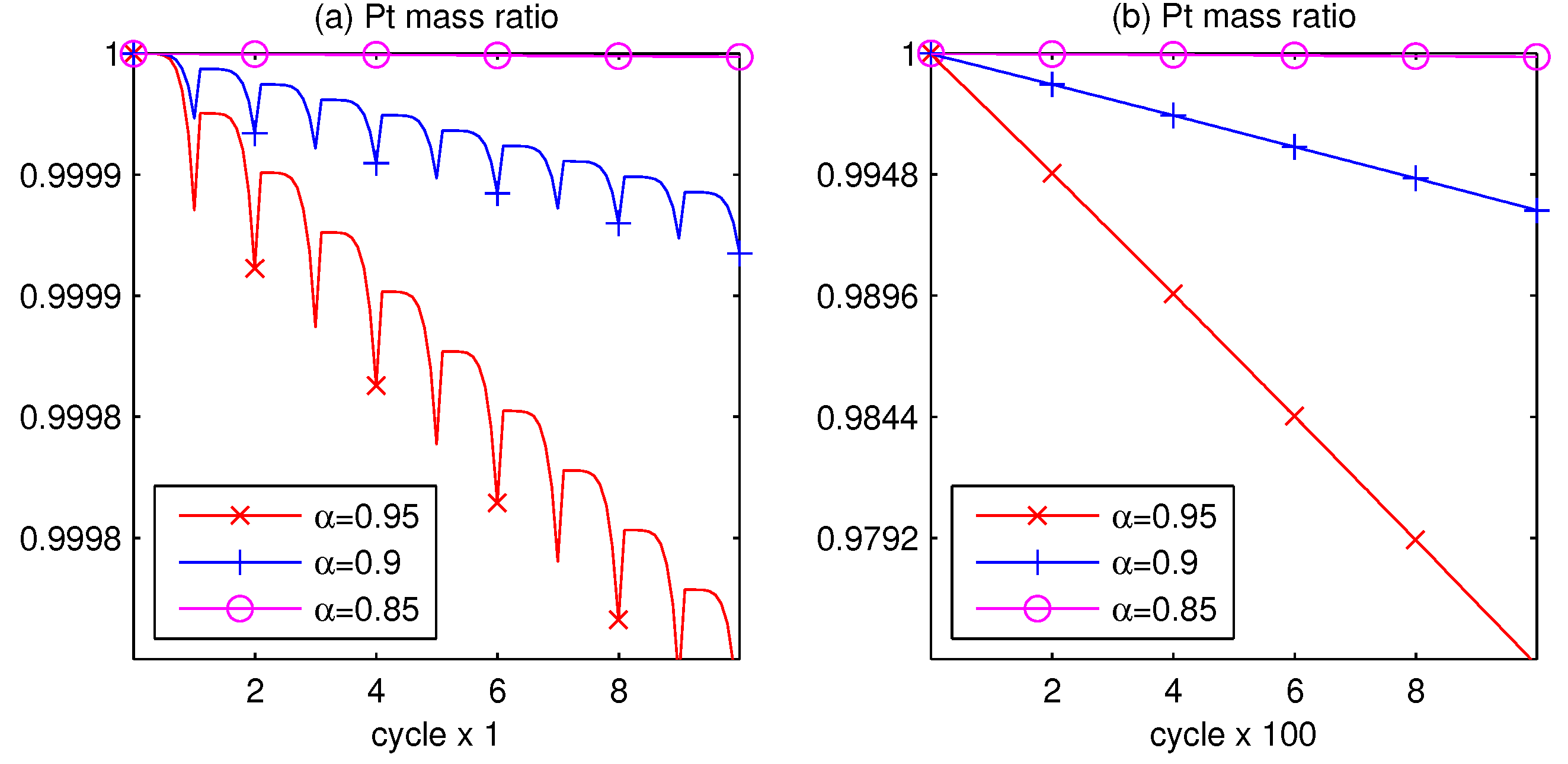
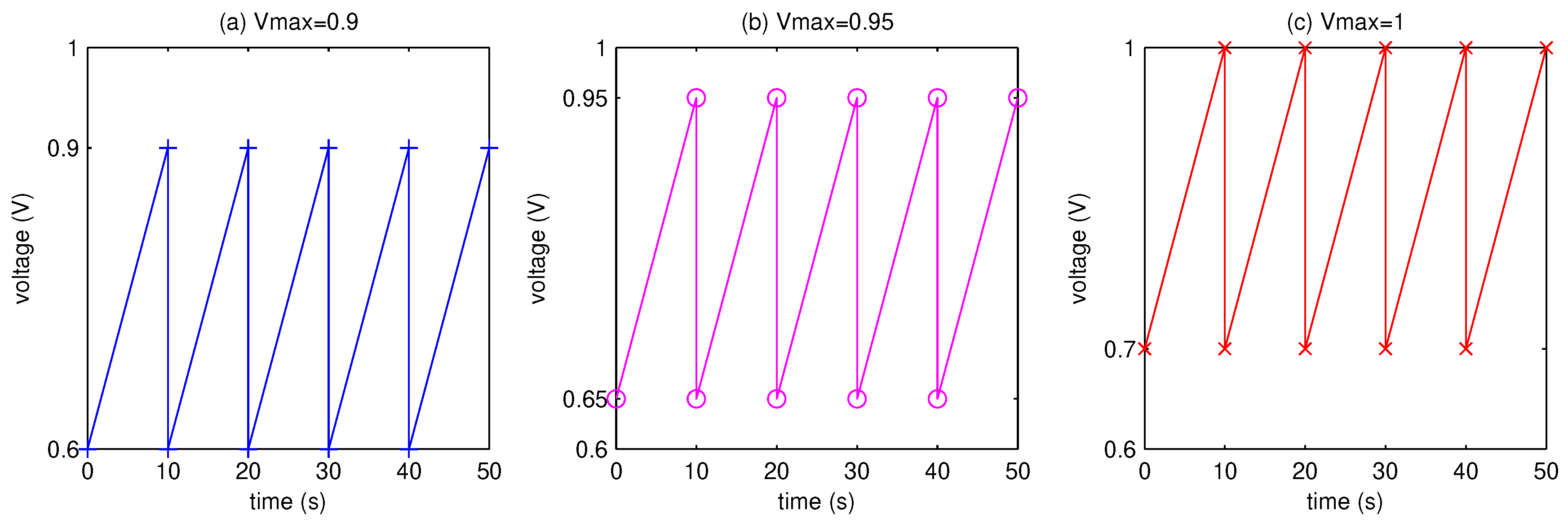
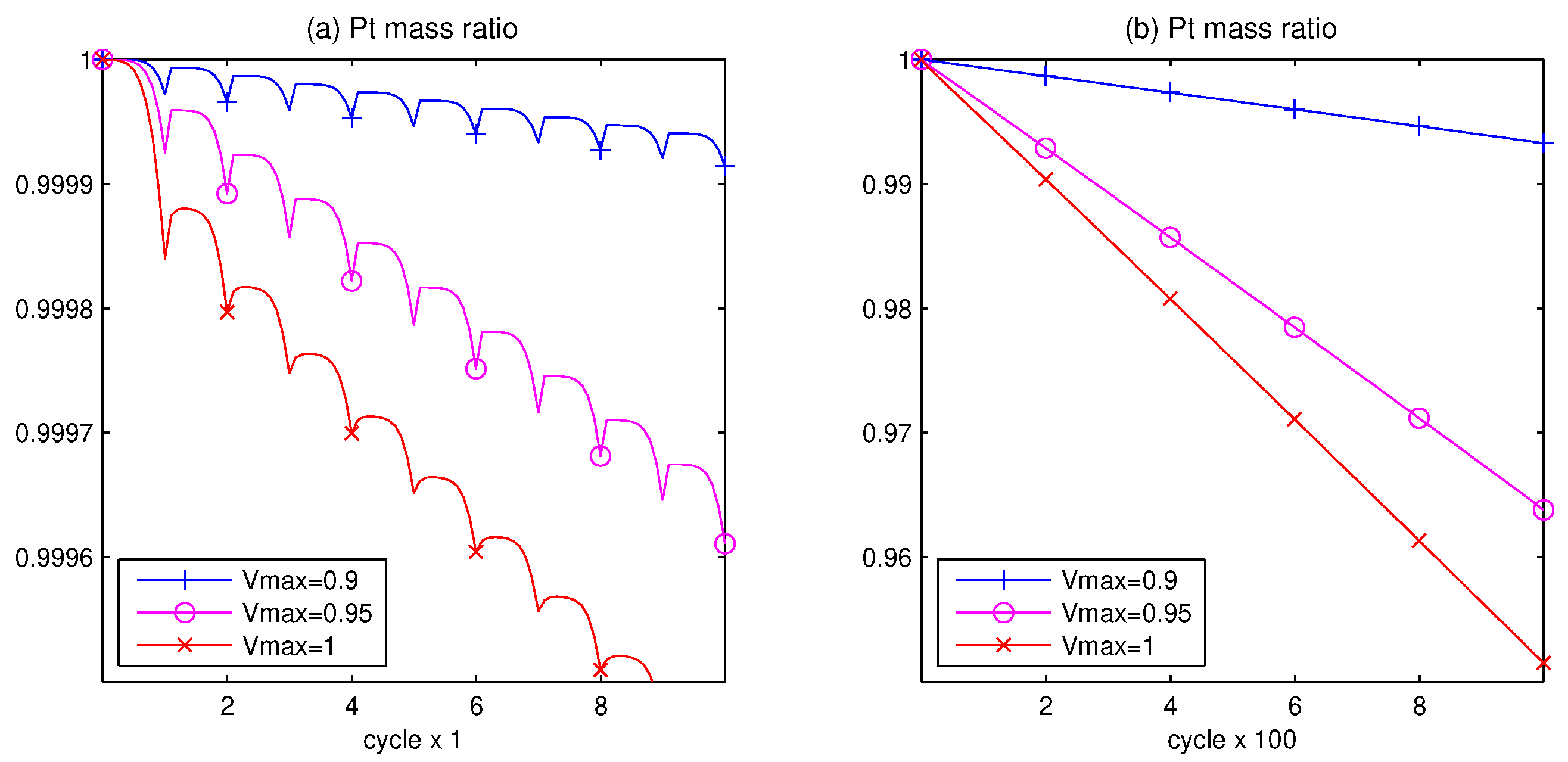
3.1. Pt Mass Loss
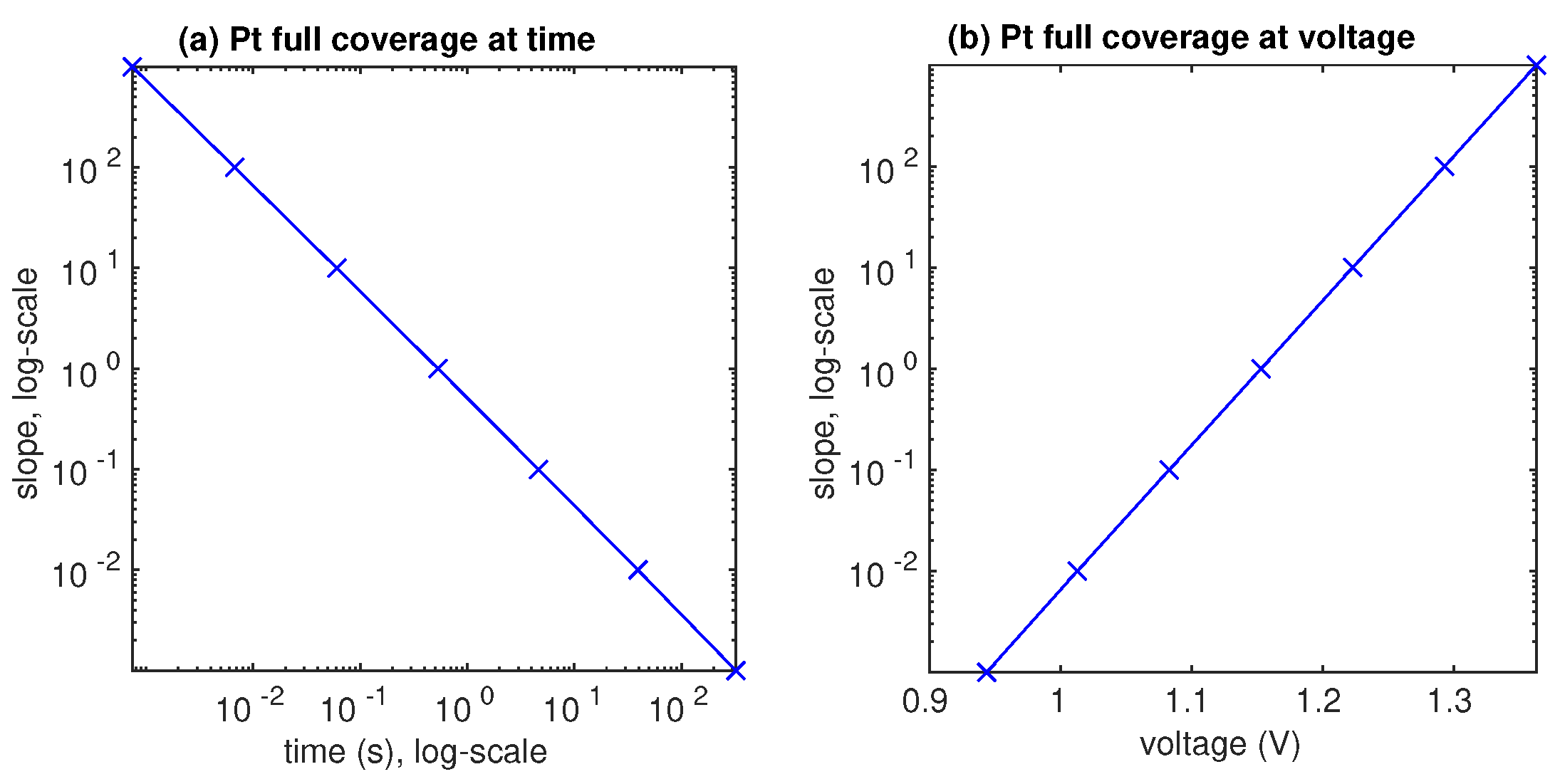
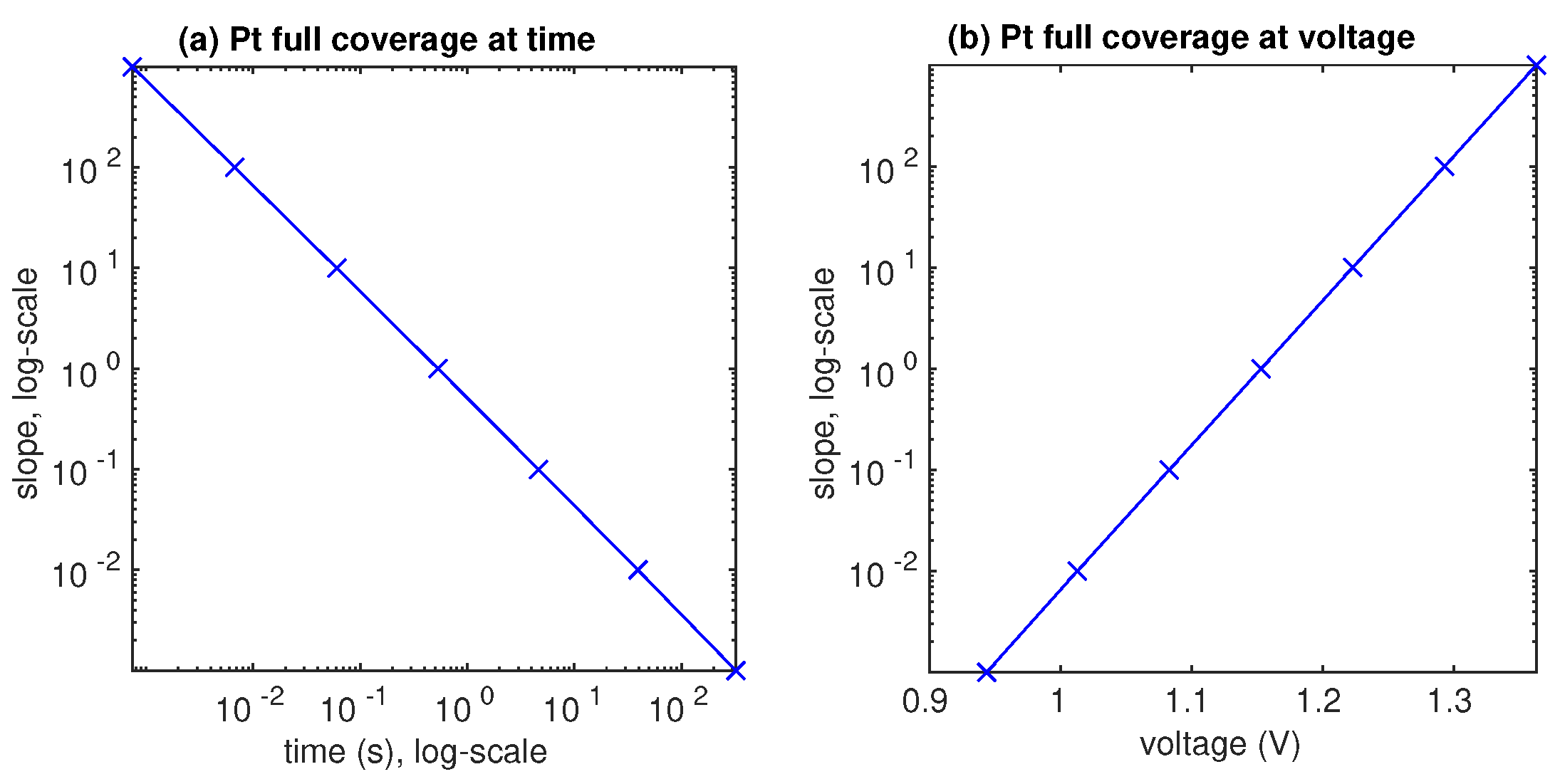
3.2. Lifetime Prognosis for CL
4. Conclusions
- The AST simulation shows that larger slope affects smaller critical time and larger critical voltage when Pt surface of the catalyst layer is completely covered by PtO.
- The computer tests demonstrate that the higher the potential difference, as well as the higher the upper potential level, the stronger the degradation phenomenon.
- Since the Pt mass loss is linear, its extrapolation from finite cycles to the full coverage, when CL is blocked, gives a prognosis for the catalyst lifetime.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AST | accelerated stress test |
| C | carbon |
| CL | catalyst layer |
| FC | fuel cell |
| GDL | gas diffusion layer |
| ODE | ordinary differential equation |
| Pt | platinum |
| PtO | platinum oxide |
| Pt | platinum ion |
| PEM | polymer electrolyte membrane |
References
- Eikerling, M.; Kulikovsky, A. Polymer Electrolyte Fuel Cells: Physical Principles of Materials and Operation; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Hacker, V.; Mitsushima, S. Fuel Cells and Hydrogen: From Fundamentals to Applied Research; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Kulikovsky, A.; Berg, T. Positioning of a reference electrode in a PEM fuel cell. J. Electrochem. Soc. 2011, 162, F843–F848. [Google Scholar] [CrossRef]
- Burlatsky, S.F.; Gummalla, M.; Atrazhev, V.V.; Dmitriev, D.V.; Kuzminyh, N.Y.; Erikhman, N.S. The dynamics of platinum precipitation in an ion exchange membrane. J. Electrochem. Soc. 2011, 158, B322–B330. [Google Scholar] [CrossRef]
- Karpenko-Jereb, L.; Sternig, C.; Fink, C.; Tatschl, R. Membrane degradation model for 3D CFD analysis of fuel cell performance as a function of time. International Journal of Hydrogen Energy 2016, 11, 13644–13656. [Google Scholar] [CrossRef] [Green Version]
- Fellner, K.; Kovtunenko, V.A. A singularly perturbed nonlinear Poisson–Boltzmann equation: Uniform and super-asymptotic expansions. Math. Meth. Appl. Sci. 2015, 38, 3575–3586. [Google Scholar] [CrossRef]
- Fellner, K.; Kovtunenko, V.A. A discontinuous Poisson–Boltzmann equation with interfacial transfer: Homogenisation and residual error estimate. Appl. Anal. 2016, 95, 2661–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González Granada, J.R.; Kovtunenko, V.A. Entropy method for generalized Poisson–Nernst–Planck equations. Anal. Math. Phys. 2018, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Kovtunenko, V.A.; Zubkova, A.V. On generalized Poisson–Nernst–Planck equations with inhomogeneous boundary conditions: A-priori estimates and stability. Math. Meth. Appl. Sci. 2017, 40, 2284–2299. [Google Scholar] [CrossRef]
- Kovtunenko, V.A.; Zubkova, A.V. Mathematical modeling of a discontinuous solution of the generalized Poisson–Nernst–Planck problem in a two-phase medium. Kinet. Relat. Mod. 2018, 11, 119–135. [Google Scholar] [CrossRef] [Green Version]
- Kovtunenko, V.A.; Reichelt, S.; Zubkova, A.V. Corrector estimates in homogenization of a nonlinear transmission problem for diffusion equations in connected domains. Math. Meth. Appl. Sci. 2020, 43, 1838–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtunenko, V.A.; Zubkova, A.V. Homogenization of the generalized Poisson–Nernst–Planck problem in a two-phase medium: Correctors and estimates. Appl. Anal. 2021, 100, 253–274. [Google Scholar] [CrossRef] [Green Version]
- Khludnev, A.M.; Kovtunenko, V.A. Analysis of Cracks in Solids; WIT-Press: Boston, MA, USA, 2000. [Google Scholar]
- Holby, E.F.; Morgan, D. Application of Pt nanoparticle dissolution and oxidation modeling to understanding degradation in PEM fuel cells. J. Electrochem. Soc. 2012, 159, B578–B591. [Google Scholar] [CrossRef]
- Kovtunenko, V.A.; Karpenko-Jereb, L. Study of voltage cycling conditions on Pt oxidation and dissolution in polymer electrolyte fuel cells. J. Power Sources 2021, 493, 229693. [Google Scholar] [CrossRef]
- Li, Y.; Moriyama, K.; Gu, W.; Arisetty, S.; Wang, C.Y. A one-dimensional Pt degradation model for polymer electrolyte fuel cells. J. Electrochem. Soc. 2015, 162, F834–F842. [Google Scholar] [CrossRef] [Green Version]
- Stariha, S.; Macauley, N.; Sneed, B.T.; Langlois, D.; More, K.L.; Mukundan, R.; Borup, R.L. Recent advances in catalyst accelerated stress tests for polymer electrolyte membrane fuel cells. J. Electrochem. Soc. 2018, 165, F492–F501. [Google Scholar] [CrossRef]
- Marcu, A.; Toth, G.; Behm, R.J. Electrochemical test procedures for accelerated evaluation of fuel cell cathode catalyst degradation. Fuel Cells 2014, 14, 378–385. [Google Scholar] [CrossRef]
- Sugawara, Y.; Okayasu, T.; Yadav, A.P.; Nishikata, A.; Tsuru, T. Dissolution mechanism of platinum in sulfuric acid solution. J. Electrochem. Soc. 2012, 159, F779–F786. [Google Scholar] [CrossRef]
- Takei, C.; Kakinuma, K.; Kawashima, K.; Tashiro, K.; Watanabe, M.; Uchida, M. Load cycle durability of a graphitized carbon black-supported platinum catalyst in polymer electrolyte fuel cell cathodes. J. Power Sources 2016, 324, 729–737. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Value | Units | Description |
|---|---|---|---|
| Hz | Dissolution attempt frequency | ||
| Hz | Backward dissolution rate factor | ||
| 0.5 | Butler–Volmer transfer coefficient for Pt dissolution | ||
| n | 2 | Electrons transferred during Pt dissolution | |
| 1.18 | V | Pt dissolution bulk equilibrium voltage | |
| 9.09 | cm/mol | Molar volume of Pt | |
| J/cm | Pt [1 1 1] surface tension | ||
| 1 | mol/cm | Reference Pt ion concentration | |
| J/mol | Fit Pt dissolution activation enthalpy | ||
| cm/s | Diffusion coefficient of Pt ion in the membrane |
| Symbol | Value | Units | Description |
|---|---|---|---|
| 0 | Potential of hydrogen ions (protons) | ||
| Hz | Forward Pt oxide formation rate constant | ||
| Hz | Backward Pt oxide formation rate constant | ||
| mol/cm | Pt surface site density | ||
| 0.5 | Butler–Volmer transfer coefficient for PtO formation | ||
| 2 | Electrons transferred during Pt oxide formation | ||
| 0.8 | V | Pt oxide formation bulk equilibrium voltage | |
| J/mol | Pt oxide dependent kinetic barrier constant | ||
| J/mol | Pt oxide–oxide interaction energy | ||
| J/mol | Fit partial molar oxide formation activation enthalpy |
| (V) | (V) | (V/s) | Rate of Pt Mass Loss | Prognosis (# Cycles) | Lifetime (h) |
|---|---|---|---|---|---|
| 0.6 | 0.85 | 0.025 | 0.999999 | 7.18527 | 19,959 |
| 0.6 | 0.9 | 0.03 | 0.993257 | 148,304 | 412 |
| 0.6 | 0.95 | 0.035 | 0.973704 | 38,028 | 106 |
| 0.65 | 0.95 | 0.03 | 0.963806 | 27,629 | 97 |
| 0.7 | 1 | 0.03 | 0.951494 | 20,616 | 57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovtunenko, V.A.; Karpenko-Jereb, L. Lifetime of Catalyst under Voltage Cycling in Polymer Electrolyte Fuel Cell Due to Platinum Oxidation and Dissolution. Technologies 2021, 9, 80. https://doi.org/10.3390/technologies9040080
Kovtunenko VA, Karpenko-Jereb L. Lifetime of Catalyst under Voltage Cycling in Polymer Electrolyte Fuel Cell Due to Platinum Oxidation and Dissolution. Technologies. 2021; 9(4):80. https://doi.org/10.3390/technologies9040080
Chicago/Turabian StyleKovtunenko, Victor A., and Larisa Karpenko-Jereb. 2021. "Lifetime of Catalyst under Voltage Cycling in Polymer Electrolyte Fuel Cell Due to Platinum Oxidation and Dissolution" Technologies 9, no. 4: 80. https://doi.org/10.3390/technologies9040080
APA StyleKovtunenko, V. A., & Karpenko-Jereb, L. (2021). Lifetime of Catalyst under Voltage Cycling in Polymer Electrolyte Fuel Cell Due to Platinum Oxidation and Dissolution. Technologies, 9(4), 80. https://doi.org/10.3390/technologies9040080