Abstract
This study investigates the synthesis and application of composite electrospun fibers incorporating coal tar pitch (CTP) and various nanomaterial additives, with a specific focus on their potential for eco-bio-applications. The research underscores the environmentally viable aspects of CTP following a thermal treatment process that eliminates volatile components and sulfur, rendering it amenable for fiber electrospinning and subsequent carbonization. Composite fibers were fabricated by integrating CTP with nanomaterials, including nickel oxide (NiO), titanium dioxide (TiO2), activated carbon (AC), and magnetite (Fe3O4). The C/NiO composite fibers exhibit notable acetone sensing capabilities, specifically displaying a rapid response time of 40.6 s to 100 ppm acetone at 220 °C. The C/TiO2 composite fibers exhibit a distinct “beads-on-a-string” structure and demonstrate a high efficiency of 96.13% in methylene blue decomposition, highlighting their potential for environmental remediation applications. Additionally, the C/AC composite fibers demonstrate effective adsorption properties, efficiently removing manganese (II) ions from aqueous solutions with an 88.62% efficiency, thereby suggesting their utility in water purification applications. This research employs an interdisciplinary approach by combining diverse methods, approaches, and materials, including the utilization of agricultural waste materials such as rice husks, to create composite materials with multifaceted applications. Beyond the immediate utility of the composite fibers, this study emphasizes the significance of deploying environmentally responsible materials and technologies to address pressing eco-bio-challenges.
1. Introduction
The global pursuit of environmental sustainability has led to transformative innovation. In this era, eco-bio-applications, driven by novel materials and advanced technologies, play a pivotal role in our collective effort to achieve a sustainable future. With climate change, pollution, resource depletion, and biodiversity loss as pressing issues, the demand for innovative, environmentally conscious approaches is urgent [1]. Eco-bio-applications represent a paradigm shift by seamlessly integrating ecological and biological principles with cutting-edge materials and techniques. In this context, the synthesis and use of new materials will be vital in bridging the gap between human progress and planetary preservation.
Sustainable development, as championed with the United Nations’ Sustainable Development Goals (SDGs), demands a holistic consideration of the environmental, social, and economic dimensions [2]. New technologies and materials, especially in the composite nanomaterials field, offer great potential in tackling these challenges. Composite nanomaterials within nanotechnology offer diverse shapes and combinations, elevating synthesis methodologies to new levels. This burgeoning field hinges on three key aspects: the composition and structure, fabrication methods, and various application domains of nanocomposites. Combining options within these dimensions provides a wide spectrum of tailored composite forms for diverse applications.
This paper centers on the production of composites via the electrospinning technique for eco-bio-applications. Bioecology, as a subfield of ecology, delves into the interplay between the environment and biological organisms.
Electrospinning stands out as a versatile technique capable of producing one-dimensional (1D) monocomponent and composite fibers, spanning an extensive spectrum of materials, from biopolymers [3] to metals and alloys [4]. These electrospun fibers have found applications across diverse fields, reflecting the adaptability and utility of this method. For a comprehensive exploration of electrospun fiber applications, extensive reviews are available [5,6,7,8,9]. In this context, a concise overview of research on the current use of electrospun fibers in eco-bio-applications is presented below, taking a closer look at electrospun fibers with the addition of nickel oxide, titanium oxide, activated carbon, and magnetite for environmental applications.
The composite nanofibers enhance the water purification process by effectively filtering and retaining fouling particles. A variety of nanofiber composite membranes, including carbon [10], nylon [11], polysulfone [12], cellulose [13], as well as polymers such as polyacrylonitrile (PAN) [14], polyvinylidene fluoride (PVDF) [15], polytetrafluoroethylene (PTFE) [16], and polypropylene (PP) [17] exhibit high performance in removing contaminants from water. Modifying these membranes enhances their performance and reduces fouling. Nanofibers show significant potential in the effective purification of water, positioning them as essential materials for future water treatment technologies.
Electrospun nanofiber membranes have the capacity to remove heavy metals, large particles, bacteria, viruses, and other textile debris, making them well-suited for water purification. Functionalizing these membranes with nanoparticles and additives such as activated carbon can address fouling issues by enhancing the effectiveness of nanostructures. This is due to several unique properties, including a lower volume-to-surface area ratio, where in the nanoscale, the number of atoms on the surface increases while defects decrease [18,19,20].
The application of nanofiber/activated carbon (AC) composites in water treatment processes is recognized as one of the most effective methods for removing various contaminants from aqueous solutions. Within these composites, activated carbon serves a crucial role by absorbing and retaining various contaminants due to its high active surface area and porous structure. In a study conducted by Shahrokhi-Shahraki et al. [21], activated carbon derived from tires was employed to purify water contaminated with synthetic heavy metals, including Pb2+, Cu2+, and Zn2+. The data obtained confirm that this activated carbon demonstrates significant potential for adsorbing heavy metals, with adsorption capacities of 322.5 mg/g for Pb2+, 185.2 mg/g for Cu2+, and 71.9 mg/g for Zn2+, respectively. It has also been observed that other factors such as the presence of other ions, amorphousness, crystallinity of the material, and pore size influence the adsorption capacity of sorbents. In the case of mesopores, the adsorption mechanism begins with the formation of a monolayer on the activated carbon surface, followed by pore condensation. Heavy metals are retained through ion exchange and chemical adsorption mechanisms [22]. To enhance the efficiency of water purification, an important factor is the high content of functional groups or active centers on the activated carbon surface, capable of binding heavy metals. Furthermore, the presence of cations like K+, Ca2+, and Mg2+ in activated carbon, which can exchange with toxic metal ions, further promotes heavy metal adsorption through ion exchange mechanisms [21].
In addition to adsorption methods for purifying water from contaminants, photocatalytic decomposition is regarded as a promising technique. Among the myriad of purification technologies, photocatalysis has proven to be a powerful approach and titanium dioxide (TiO2) stands out as a leading photocatalyst due to its non-toxicity, exceptional photoactivity, and low cost [23]. However, despite TiO2’s remarkable photocatalytic capabilities, its full potential is often hindered by several limitations, such as the rapid recombination of photogenerated electron–hole pairs and the restricted light absorption in the ultraviolet (UV) range [24]. To overcome these hurdles and enhance TiO2’s photocatalytic performance, the incorporation of carbon nanofibers has emerged as a transformative solution. TiO2/carbon nanofiber composites have garnered substantial attention in recent years for their ability to synergize the merits of TiO2 and carbon-based materials, effectively addressing the shortcomings of traditional TiO2 photocatalysts. Due to this, most of the researchers have implemented electrospinning technology [25,26,27], which has demonstrated outstanding outcomes in the treatment of wastewater containing different kinds of dyes.
In reference [28], an excellent example of using a combination of TiO2 and metal–organic frameworks, specifically zeolitic imidazolate framework-8 (ZIF-8), is presented. This combination was prepared using ultrasonic and electrospinning methods, demonstrating good photoactivity and the ability to maintain its performance over multiple cycles (88.7% efficiency after five cycles). The use of multilayer composites, including TiO2, carbon nanotubes, and polymethyl methacrylate, created using the same electrospinning method, can enhance the photocatalysis effect for a broader range of dyes, expanding its application scope [29]. Furthermore, a novel photocatalyst [30], known as Janus nanofiber heterojunction photocatalysts (JNHPs), was developed. JNHP incorporates a TiO2/C nanofiber side that responds to UV light and a Bi2WO6/C nanofiber side that is responsive to visible light, efficiently utilizing sunlight. JNHP excels in hydrogen production and methylene blue degradation without the need for noble metals as co-catalysts. This approach holds promise for the development of other bifunctional nanofiber photocatalysts.
Composite nanomaterials find critical applications in the detection of hazardous gases, including toxic, flammable, and explosive substances [31,32,33,34]. Recent interest has centered on gas sensors utilizing carbon fibers and metal oxide nanoparticles, particularly NiO, renowned for their simple production methods and high selectivity [35].
NiO-based gas sensors operate by measuring resistance changes in the sensing layer upon gas molecule adsorption. Nanofibers, with their exceptional surface-to-volume ratio, high porosity, and enhanced active centers, improve gas adsorption, enhancing gas detection performance [36]. Among various nanofiber fabrication methods, electrospinning stands out as a promising, cost-effective, and versatile technique for producing NiO-based composite nanofibers. These nanofibers enhance electrical transport, specific surface area, and contact area with the target gas, and reduce charge transfer time, making them highly suitable for gas sensor development.
In [37], PAN/NiO-based gas sensors were successfully used for methanol detection, demonstrating high selectivity and stability. Gas sensors not only find application in environmental protection but also in biomedicine. For instance, poly(vinyl alcohol) (PVA)/NiO nanofibers produced using electrospinning exhibited a rapid response and recovery time, making them suitable for formaldehyde vapor detection [38]. Electrospun NiO nanograins can be utilized in exhaled air sensors for lung disease diagnoses [39]. Incorporating electrospun nanomaterials into gas sensors results in ultrahigh sensitivity, excellent selectivity, long-term stability, and cost-effectiveness even in challenging conditions.
The potential utilization of magnetic composite fibers, particularly those incorporating magnetite nanoparticles, is determined with their physicochemical and magnetic characteristics. These properties make them remarkably versatile and applicable in a wide range of fields, including sensing, biomedicine, electronics, telecommunications, environmental protection, and energy storage, as discussed in references [40,41]. For example, composite fibers containing magnetite nanoparticles exhibit a significant level of magnetization, a crucial characteristic that underscores their usefulness [42]. It is worth noting that the inclusion of magnetite nanoparticles not only enhances the dielectric properties and magnetic characteristics of the fibers but also exerts influence over their orientation and size. This study also investigated the effects of magnetic fields on fiber orientation during the electrospinning process, resulting in reduced fiber dimensions and improved fiber alignment [43].
In references [44,45], composite fibers based on magnetite nanoparticles in poly(ethylene oxide) (PEO) and polydopamine (PDA) matrices were fabricated through electrospinning for the removal of Pb(II) ions from solutions. Reference [46] discusses the production of polyvinylpyrrolidone (PVP)/Fe3O4 fibers for drug delivery as drug-loaded magnetic materials. Additionally, PVA/Fe3O4 fibers, as described in [47], can be used for physisorption, and they also serve as biomembranes, as discussed in [48]. Alginate/Fe3O4 electrospun fibers, as demonstrated in reference [49], find applications in adsorption and separation of antibiotics, including the separation of ciprofloxacin hydrochloride from aqueous solutions. As a result, the growing body of research dedicated to the development of functional magnetic fibers, distinguished by their exceptional magnetic properties attributed to the presence of magnetite nanoparticles, is indeed significant.
This article presents the results obtained by the Institute of Combustion Problems research group regarding the synthesis of composite materials using the electrospinning method and their environmental applications. The study involved the preparation of carbon-based composite fibers with the addition of MOx (NiO, TiO2, and Fe3O4), as well as carbon–carbon fibers through the incorporation of activated carbon particles. The resulting fibers were subject to testing for the purification of water from metal ions and organic pollutants through adsorption and photocatalytic decomposition. Additionally, the fibers were evaluated for their capability to detect toxic gases in the atmosphere. One noteworthy aspect of this study is the use of coal tar pitch in conjunction with the primary PAN solution as an alternative, cost-effective precursor. Furthermore, the production of activated carbon is involved in the use of agricultural waste, specifically rice husk. This illustrates the potential for converting industrial and agricultural waste materials into environmentally protective materials.
2. Materials and Methods
This investigation involves multiple experimental phases, which can be categorized into three primary stages:
- Synthesis of basic components intended for subsequent composite material fabrication.
- Synthesis of composite fibers through electrospinning and their subsequent heat treatment.
- Evaluation and practical implementation of the resulting materials.
It is important to note that each stage incorporates fundamental techniques for examining intermediate and final products, including morphological and physicochemical analyses. Some of these steps are illustrated in Figure 1.
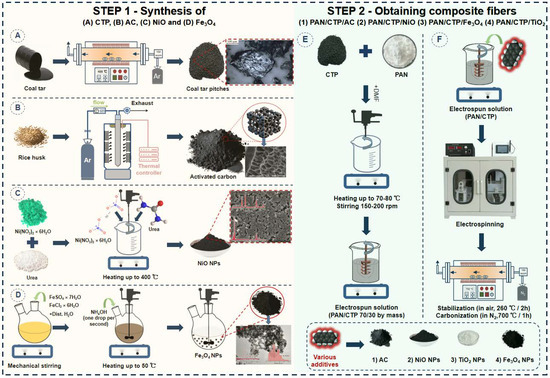
Figure 1.
The key stages involved in the production of various composite fibers. (A)—Synthesis of coal tar pitches from coal tar by heat treatment; (B)—Synthesis of activated carbon from rice husk; (C)—Solution-combustion synthesis of nickel oxide nanoparticles; (D)—Synthesis of magnetite nanoparticles by chemical condensation; (E)—Preparation of electroforming solution based on PAN and CTP; (F)—Preparation of PAN/CTP-based carbon fibres incorporating diverse additives such as activated carbon, NiO, TiO2 and Fe3O4.
2.1. Production of Coal Tar Pitches (CTPs) with Thermal Treatment
Coal tar pitches were obtained through the thermal conversion of coal tar in a tube furnace equipped with a quartz reactor under an inert atmosphere. To investigate the influence of temperature on pitch formation, heat treatment was carried out within the temperature range of 200–500 °C, while maintaining an argon gas flow rate of 90–92 cm3/min. A pre-dried and accurately weighed porcelain or quartz boat was loaded with coal tar, and its initial mass was recorded for a subsequent mass loss analysis. The boat containing the coal tar was then placed into the reactor and subjected to argon purging to eliminate atmospheric air. Argon purging was maintained for 5 min, after which the reactor was gradually heated to the specified temperature range (200–500 °C). The temperature treatment duration was set at 1 h. Once the heat treatment was completed, the reactor heating was turned off, and the sample was allowed to cool to ambient temperature within an argon-filled environment. After extraction, the product boat was re-weighed to measure the mass loss. Subsequently, the resulting material was used for electrospinning to produce fibers.
2.2. Synthesis of C/NiO Composite Fibers
Nickel oxide nanoparticles were synthesized using the solution combustion method. Nickel nitrate hexahydrate (Ni(NO3)2∙6H2O) was employed as the oxidizer, and urea (NH2CONH2) as the fuel. The process commenced by dissolving these reagents in distilled water and evaporating the solution to a final volume of 5–7 mL, resulting in a gel-like substance as most of the water evaporated. Subsequently, the mixture was heated to 260 °C, causing spontaneous ignition. The choice of ignition temperature was based on urea’s decomposition point, which can reach temperatures of up to 1200 °C in the burning zone. This led to the deposition of the final product on the inner surfaces of the glassware, yielding a fine, jet-black powder.
To prepare a fiber-forming PAN and CTP solution with nickel oxide nanoparticles, the following steps were followed:
- A well-suited suspension of powdered PAN was combined with N,N-dimethylformamide (DMF) in a mass ratio of 9:91. This mixture was stirred on a magnetic stirrer at 150 rpm and maintained within a temperature range of 70–80 °C for 2 h until a homogeneous solution was achieved.
- Once complete homogenization was achieved, the PAN solution was blended with small quantities of CTP in a mass ratio of 70:30. The resulting mixture underwent ultrasonic treatment in a bath operating at a temperature range of 70–80 °C for 60 min, ensuring the uniform dispersion of CTP within the polymer solution.
- Subsequently, nickel oxide powder was added to the solution, maintaining a weight ratio of 70:30 ((PAN/CTP):NiO). This suspension was continuously stirred on a magnetic stirrer for a duration of 12 h.
The electrospinning process was conducted using a specialized apparatus consisting of a solution delivery system, a syringe reservoir equipped with a metal needle, a power source, and a variable-speed drum collector. The fiber formation procedure was carried out under ambient conditions, with a temperature range of 20–30 °C and a relative humidity of 30–40%.
The polymer solution was loaded into a 5 mL syringe, with a metal needle securely attached to its end. A negative voltage was applied to the needle, while a positive voltage was applied to the collector. Specifically, both the needle and the drum collector were subjected to a voltage of 15 kV. The solution feed rate was maintained at 1.0 mL/h, the drum collector had a radius of 20 cm, and the distance between the needle and the collector was set at 15 cm. To facilitate fiber deposition, the collector’s surface was lined with aluminum foil. Voltage levels and the distance between the electrode and the collector were meticulously adjusted to ensure the stable formation of the electrospinning jet. Subsequently, the fibers underwent a two-step process involving stabilization and carbonization.
For stabilization, the PAN/CTP/NiO fibers were placed in a tubular CVD (Chemical Vapor Deposition) furnace with a quartz tube, heated to 260 °C, and kept at this temperature for 1 h in an air atmosphere. Afterward, the furnace was turned off, and the sample was allowed to cool to room temperature inside the furnace.
Subsequently, carbonization was carried out in the same CVD furnace. The quartz tube was purged with high-purity nitrogen (99.993%) to eliminate any traces of air and prevent contact with oxygen. The furnace was then heated to 700 °C, and the carbonization process lasted for 1 h. After completion, the furnace was turned off, and the sample was cooled to room temperature in a nitrogen atmosphere, again without removal from the reactor. The obtained sample was named “C/NiO fibers”.
To assess the gas sensitivity, the C/NiO fibers were meticulously ground in an agate mortar. The ground composite fibers were blended with PVDF at a mass ratio of 85:15 (C/NiO:PVDF), with PVDF serving as a binding agent in the mixture. The resulting viscous suspension was applied onto a microchip specially designed with interdigitated gold nanowires and two electrodes mounted on aluminum oxide. Subsequent to the application of the suspension, the sample was subjected to a 2 h drying process in an oven set at a temperature of 100 °C.
2.3. Synthesis of C/TiO2 Composite Fibers
The production of nanostructured composite fibers, incorporating PAN and CTP with the addition of titanium oxide nanoparticles, was successfully achieved. The preparation of the fiber-forming solution closely adhered to the methodology outlined in a previous section of this article. In brief, CTP was added to a 9% PAN:DMF solution with a PAN:CTP weight ratio of 7:3. Subsequently, titanium oxide nanoparticles were introduced, maintaining a (PAN/CTP):TiO2 weight ratio of 7:3. The electroforming of fibers was carried out, followed by the processes of stabilization and carbonization. The samples obtained after carbonization were designated as “C/TiO2 fibers”.
The investigation of the photocatalytic performance of carbon nanofibers modified using titanium dioxide (C/TiO2) was carried out through the photocatalytic degradation of methylene blue (MB). This degradation process occurred under the irradiation of a 300 W xenon lamp, with experiments conducted at room temperature. The lamp was positioned approximately 15~20 cm above the solution surface.
In the experimental procedure, 5 mg of C/TiO2 were dispersed within a 50 mL solution of methylene blue, initially having a concentration of 10 mg/L. Following the introduction of the nanofibers, the mixed solution was allowed to stand in the dark and underwent stirring for 30 min to establish adsorption–desorption equilibrium within the system.
Subsequently, the entire system was placed under the xenon lamp to initiate the photocatalytic degradation process. At 10 min intervals, 5 mL aliquots of the solution were extracted and subjected to centrifugation. The purpose of this centrifugation step was to separate solid residues from the liquid phase for a further analysis.
2.4. Synthesis of C/Fe3O4 Composite Fibers
Magnetite (Fe3O4) nanoparticles were synthesized using a chemical condensation method as previously detailed in reference [50]. In brief, an aqueous solution of iron (III) chloride with a concentration of 0.32 mol/L and iron sulfate with a concentration of 0.2 mol/L was combined in a heat-resistant flask. This mixture was agitated using a heated magnetic stirrer, while a 25% aqueous ammonia solution was added gradually, at a rate of one drop per second. The temperature of the iron salt solution was maintained at 50 °C. Subsequently, magnetite nanoparticles were extracted from the solution using a permanent magnet, washed several times with water to neutralize the reaction, and then dried under standard conditions.
The fabrication of nanostructured composite fibers, incorporating PAN and CTP with the inclusion of magnetite nanoparticles, was successfully achieved. The preparation of the fiber-forming solution closely adhered to the procedure described in a previous section. In summary, CTP was incorporated into a 9% PAN/DMF solution with a PAN/CTP weight ratio of 7:3. Following this, magnetite nanoparticles were added, maintaining a PAN/CTP:Fe3O4 weight ratio of 7:3. Subsequently, the electroforming of fibers was conducted, followed by the crucial stages of stabilization and carbonization. The resulting samples were designated as “C/Fe3O4 fibers”.
2.5. Synthesis of C/AC Composite Fibers
The highly porous activated carbon (AC) was produced from rice husk waste, following the methodology detailed in a prior investigation [51]. The fabrication of composite fibers, incorporating PAN and CTP with the inclusion of activated carbon, was successfully carried out. The preparation of the fiber-forming solution closely adhered to the methods described in previous sections. In summary, CTP was introduced into a 9% PAN:DMF solution with a PAN:CTP weight ratio of 7:3. Subsequently, activated carbon was added, maintaining a (PAN/CTP):AC weight ratio of 7:3. Subsequently, the electroforming of fibers was performed, followed by the crucial stages of stabilization and carbonization. The samples obtained after carbonization were designated as “C/AC fibers”.
The sorption characteristics of the resulting C/AC composite fibers regarding the adsorption of manganese (II) ions from aqueous solutions were investigated with respect to adsorption time. Calibration solutions were carefully prepared to contain manganese (II) ions at concentrations of 5, 10, 15, and 20 mg/L, following the guidelines of State Standard, GSO (Certified Reference Material) no. 7875-2000. The working concentration was set at 10 mg/L. Samples of C/AC fibers, weighing 0.25 g, were immersed in 200 mL beakers filled with 100 mL of the working solution. At specific time intervals (3, 10, 30, 60, and 720 min), samples were retrieved from the beakers to determine the manganese ion content using atomic absorption spectrometry.
2.6. Methods of Characterization
2.6.1. X-ray Phase Analysis
X-ray patterns were obtained with a DRON-4 universal X-ray diffractometer equipped with an IBMPC-based digital control and registration system using copper radiation. This method allowed us to accurately determine the lattice parameters of crystalline materials and perform qualitative and quantitative phase analyses of materials. The X-ray tube operated at voltages up to 30 kV, with a tube current of 30 mA. The goniometer moved in steps of 0.05° 2θ, and the measuring point intensity reached up to 1.0. The sample’s own-plane rotation speed was 60 rpm.
X-ray data were processed to determine angular position and reflection intensity using the ‘Fpeak’ program. The phase analysis was conducted through the ‘PCPDFWIN’ program, which utilized a diffractometric database. Spectra obtained were identified with reference to the JCPDS X-ray database. Notably, the primary hardware error in the X-ray counting measurement did not exceed 0.4%.
2.6.2. Electron Microscopy
The structure, size, and morphology of the obtained samples were investigated using a Quanta 200i 3D scanning electron microscope (FEI, Hillsboro, OR, USA) with an accelerating voltage of 30 kV. This microscopy analysis was carried out at the “National Nanotechnology Laboratory of Open Type” within KazNU (Al-Farabi University, Almaty, Kazakhstan). This method allowed us to determine surface structures in images, facilitating the assessment of both structural characteristics and the sizing of individual particles. Notably, the Quanta 200i 3D SEM is equipped with an energy-dispersive X-ray system (EDS), which enables the analysis of a wide range of chemical elements, spanning from B to U. The energy resolution of this system is 132 eV (Mn Kα).
In addition, the structural and morphological characteristics of magnetite nanoparticles were investigated using a transmission electron microscope, JEM-1011 (JEOL, Tokyo, Japan) in the Kazakhstan-Japan Innovation Center of the Kazakh National Agrarian Research University. This microscope is equipped with a digital camera, Morada (Olympus, Tokyo, Japan). Key technical specifications include an adjustable accelerating voltage within the range of 40 to 100 kV, a dot resolution of 0.3 nm, a line resolution of 0.14 nm, a LaB6 electron gun, and a magnification range from 100 to 1,000,000.
Fiber diameters and particle sizes were determined from a set of SEM/TEM images using the open-source program “ImageJ”, specially developed for an image analysis and processing, which automates the calculation of the average particle diameter and standard deviation.
2.6.3. Raman Spectroscopy
Raman spectra of the samples were obtained on an NT-MDT NTegra Spectra spectrometer located at the “National Nanotechnology Laboratory of Open Type” of KazNU (Al-Farabi University, Almaty, Kazakhstan).
Raman spectroscopy was carried out with unpolarized radiation from a semiconductor diode laser as the excitation source, emitting at a wavelength of λexc = 473 nm. The wavenumber scale had a relative error tolerance maintained within ±0.5%.
2.6.4. P-45X Potentiostat-Galvanostat Workstation
The gas-sensitive properties were evaluated using a static test method. The sensor was positioned within a 10 L test chamber, with its electrodes connected to a P-45X potentiostat-galvanostat workstation. Current characteristics between the two electrodes were measured while exposing the sample to the analyzed gas. This procedure enabled the assessment of the composite fibers’ sensitivity to gases when connected to the specialized microchip and measurement equipment.
A volume of vapor saturated with acetone was introduced into the chamber to create an environment with a specific concentration of acetone, and the gas detection characteristics of the sensor were recorded. The sensor response was determined using the following Formula (1):
where Ia is the sensor current in air; Ig is the sensor current in the acetone environment.
2.6.5. UV-Vis Spectroscopy
The UV-Vis absorption spectrum of the material is to analyze the change in UV-Vis spectra of methylene blue solutions before and after the photocatalytic degradation process after centrifugation.
The analysis was conducted using a UV-Vis spectrophotometer, enabling us to quantify the amount of methylene blue adsorbed using the material based on the observed spectral changes.
This technique provided valuable insights into the adsorption capacity and efficiency of the test material for methylene blue molecules. The spectral analysis yielded essential data concerning the degradation efficiency of methylene blue over time, facilitating an evaluation of the photocatalytic activities of the C/TiO2.
2.6.6. Atomic Absorption Spectroscopy
Atomic absorption spectroscopy was used to evaluate the adsorption capacity of the obtained materials on metal ions. Alterations in the concentration of manganese ions in acidified aqueous solutions were analyzed using atomic absorption spectra, utilizing a “PerkinElmer AAnalyst 200” instrument. Sample concentrations were determined by introducing the sample into the flame of an acetylene-air burner as a nebulizer. The margin of error associated with the experiment was within ±2.5%.
where C0 and C are concentrations of metal ions in the solution before and after precipitation, mg/L; V is the volume of the solution, L; g is the mass of the composite fibers, g.
3. Results and Discussion
3.1. Characterization of Coal Tar Pitches Obtained with Thermal Treatment
The coal tar used in this study was sourced from the Shubrakol coal deposit in Kazakhstan. The Shubrakol field holds substantial reserves, amounting to an impressive 1.5 billion tons. Coal tar itself is characterized as a dense, dark liquid with a distinctive odor, exhibiting a viscosity of 1.04 g/cm3.
To examine the properties of coal tar, specimens were subjected to controlled heating processes at various temperature intervals, and the mass variation was meticulously assessed by comparing the pre- and post-treatment weights of the samples. The resulting data is summarized in Figure 2, providing insights into the extent of mass reduction following each treatment. As indicated in the tabulated data in Figure 2, the peak of mass reduction occurs at the temperature threshold of 500 °C, representing approximately 76% of the initial mass. The application of thermal treatment accelerates the elimination of volatile constituents within the coal tar substrate.
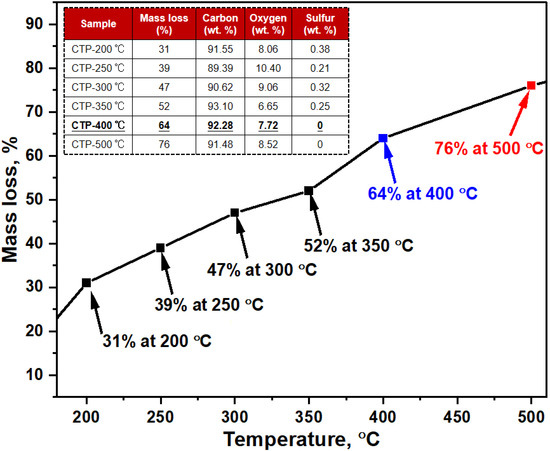
Figure 2.
Mass loss and elemental composition of coal tar after heat treatment.
The data presented on the heat treatment of coal tar are instructive in understanding its chemical transformation with increasing temperature. The significant mass loss observed at 500 °C indicates the substantial removal of volatile fractions. This is crucial for materials intended for further high-temperature applications, such as carbonization. The removal of sulfur from the composition at 400 °C is particularly significant. Sulfur can be undesirable in carbon materials as it leads to impurities and can affect the final properties of carbon-based materials.
The reduction in volatile content and the removal of sulfur at higher temperatures enhance the carbonization potential of the material, which is favorable for the production of high-quality carbon materials. The removal of volatile components implies greater structural stability at elevated temperatures, which is beneficial for applications requiring thermal resistance.
The analysis of SEM imagery reveals significant structural alterations in coal tar samples subjected to varying processing temperatures. At an initial processing temperature of 200–250 °C, discernible transformations manifest in the form of a porous microstructure and the initiation of active mesophase centers. Subsequent elevation of the processing temperature to 300 °C results in a pronounced escalation in surface degradation. The treated sample exhibits a more pronounced topographical relief, accompanied with a heightened density of mesophase centers per unit area. At a processing temperature of 350 °C, a critical transition is observed, marking the shift from an isotropic to an anisotropic structural configuration. Notably, this sample demonstrates the complete removal of volatile fractions, yielding a homogeneous surface with mesophase centers averaging around 2 µm in size.
When coal tar is processed at 400 °C, a comparable anisotropic structure forms, occasionally featuring a layered arrangement. This structural alteration accompanies an increased degree of graphitization within the sample. The size of the mesophase particles notably expands to a range of 3.5–5 µm. The culmination of this temperature-based experimentation occurs at a coal tar processing temperature of 500 °C, resulting in a definitive transition from an isotropic to an anisotropic structural state.
Figure 3 shows the Raman spectra obtained from coal tar subjected to different temperature treatments. The interpretation of these Raman spectra was carried out with reference to the comprehensive analysis presented in research article [52].
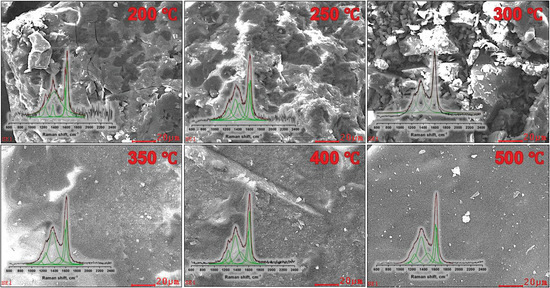
Figure 3.
SEM images (with Raman shifts) of coal tar after heat treatment.
In the context of the original graphite, two primary first-order peaks manifest themselves at wavelengths of 1355 cm−1 (referred to as the D peak, associated with the defective Raman zone) and 1575–1582 cm−1 (referred to as the G peak, attributed to the presence of carbon atoms in the sp2 state and located within the graphite lattice plane). In particular, the second-order spectral lines around ~2710 cm−1 show minimal resolution and considerable disorder, making their identification impossible. Several key parameters are crucial in interpreting Raman spectra: λD and λG, the wavelengths of the D and G peaks, respectively, measured in cm−1; ID and IG, the intensities of the D and G peaks in relative units; and R, the ratio of the intensities of the D and G peaks (ID/IG). The appearance of graphite nanocrystallites in the sample is accompanied with a shift of the G peak from 1575–1582 cm−1 to higher values, around ~1600 cm−1. Accurate interpretation of peak half-width and area requires careful determination of the background line in the Raman spectrum.
Within the samples designated CTP-200, CTP-250, and CTP-300, there is an observable shift of the G-peak towards higher frequencies, approximately in the range of ~1405–1428 cm−1. This phenomenon can be attributed to the presence of clusters with a reduced number of aromatic rings in these samples. In particular, there is considerable background noise in these two samples, which makes it difficult to accurately determine the degree of graphitization. Consequently, the R value, indicating the ID/IG ratio, has not been considered for these samples. A brief summary of the wavelengths of the G and D peaks, their corresponding intensities, and R values are given in Table 1.

Table 1.
Characteristics of G and D peaks for coal tar samples treated at different temperatures.
For sample CTP-350, a significant change in both the intensities and positions of the G and D peaks can be seen. This change implies the elimination of all volatile fractions and the onset of a transition from a disordered to a more ordered structural state, characterized by the formation of mesophase centers. Additionally, for samples CTP-350, CTP-400, and CTP-500, an observable shift of the D peak towards the region of 1600–1610 cm−1 is noted, which can be attributed to the appearance of nanocrystalline mesophase centers.
Based on the data presented in Table 1, it is possible to estimate the degree of graphitization using the equation provided [53]:
where g is the graphitization degree, %; R is the ID/IG ratio; n is the maximum value of R obtained during the study (in this case, 0.7214).
Thus, the calculation showed that for sample CTP-350, the degree of graphitization is ~5%; for sample CTP-500, it is ~10%. For sample CTP-400, this indicator cannot be determined, because the value of R was taken as the maximum value (n).
3.2. Characterization of C/NiO Composite Fibers
Within the X-ray diffraction pattern (Figure 4), there are four noticeable peaks. These peaks are distinctly located at 2θ angles of 37.27°, 43.27°, 62.84°, and 75.42°. Importantly, they correspond to cubic NiO crystalline structures, with each peak originating from a distinct diffraction plane: (111), (200), (220), and (311), respectively. It is worth noting that these diffraction peaks closely match the expected pattern defined in the JCPDS card 47-1049. Consequently, the XRD data strongly suggest that the sample primarily comprises single-phase nickel oxide. The size of the nickel oxide crystallites, estimated using the Scherrer equation, approximates 480 Å.

Figure 4.
SEM (with EDAX-analysis and XRD pattern) image of NiO nanoparticles.
The SEM analysis (Figure 4) shows that the nanoparticles exhibit a spherical morphology and, notably, tend to form agglomerates. This aggregation phenomenon is likely driven by the nanoscale dimensions of the crystallites and the presence of numerous uncompensated surface bonds. The inherent high surface energy associated with nanocrystals compels them to aggregate, a mechanism aimed at minimizing surface energy during the synthesis process.
Furthermore, the specific surface area of the NiO nanoparticles has been determined to be 65.544 m2/g, which is a promising outcome for oxide materials. In comparison, a previous study conducted by the authors [54] involved the synthesis of nickel oxide through the sol-gel method, yielding a significantly lower specific surface area of 5.8 m2/g. This comparison underscores that nickel oxide produced via the solution combustion method exhibits reduced dimensionality and heightened porosity, making it a noteworthy advancement in materials synthesis.
PAN/CTP/NiO composite fibers were fabricated via the electrospinning technique, subsequently of stabilization and carbonization processes. In Figure 5, depiction is presented, encompassing both the SEM image and the accompanying diameter distribution analysis of the electrospun composite fibers following the stages of stabilization and carbonization.
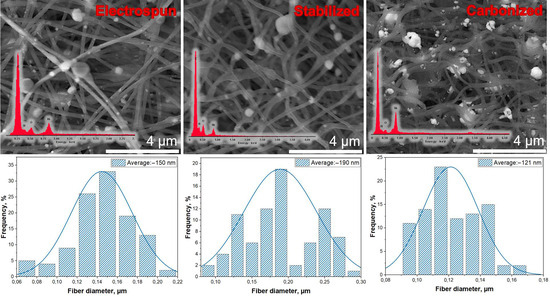
Figure 5.
SEM (with EDAX-analysis) image and fiber distribution of PAN/CTP/NiO fibers after electrospinning, stabilization, and carbonization processes.
Fibers produced via the electrospinning method exhibit structural resilience during subsequent heat treatment processes, including stabilization and carbonization. These critical steps, involving high temperatures and chemical transformations, do not compromise the integrity of the electrospun fibers. This retention of structure is vital for the practical applications.
SEM images illustrate the successful inclusion of nickel nanoparticles within the fiber structure. Additionally, these images reveal the formation of visible agglomerates on the fiber surface. NiO particles have high adhesion to the fiber surface due to chemical and physical changes occurring during the carbonization process.
The alteration of average fiber diameter throughout the fabrication process, from electrospinning (150 nm) to stabilization (190 nm) and finally to carbonization (121 nm), can be attributed to several factors. During stabilization, elevated temperatures cause some swelling due to cross-linking and the elimination of volatile components. Subsequently, carbonization results in a reduction in diameter due to the removal of non-carbon atoms and the development of a denser, graphitic carbon structure. This dynamic process allows for precise control over fiber diameter and, consequently, material properties for various applications.
To determine the optimal operating temperature for the composite fibers, an investigation was conducted into the influence of heating temperature on the response of these fibers when exposed to 100 ppm of the test gas, namely acetone (as depicted in Figure 6). Observations reveal a nuanced temperature-dependent response pattern. As the temperature escalates from 160 °C, the sensor response exhibits a gradual increase, culminating at its zenith around 220 °C, only to exhibit a rapid decline with further temperature elevation. This observed behavior primarily hinges on the temperature’s impact on the adsorbed gas.
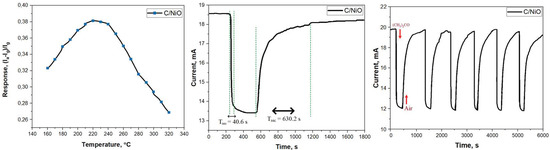
Figure 6.
Graph of resistance dependence on operating temperature (a), response and recovery time (b), and operating time (c) for C/NiO-based gas-sensitive material for acetone/air mixture.
In ambient air, oxygen molecules are adsorbed onto the surface of the sensing material. As the temperature rises, a transition occurs from physical adsorption to chemical adsorption for the adsorbed gas. Notably, oxygen ions, which facilitate electron transfer within the adsorbed gas, can significantly amplify the reaction within a specific temperature range. However, if the temperature exceeds a certain threshold, it can impede the adsorption of gas onto the sensing material’s surface.
Figure 6 illustrates the composite fibers’ response to 100 ppm of acetone at the optimal operating temperature. Upon introduction of gaseous acetone into the test chamber, the sensor current value exhibits a rapid decrease, swiftly followed by an increase in sensor current upon the cessation of acetone exposure.
Both microdispersed and nanodispersed nickel oxide particles are recognized for their gas-sensitive properties, yet their sensitivity and response times to gas exposure may diverge. Generally, nanodispersed nickel oxide particles boast a larger surface area in comparison to their microdispersed counterparts, endowing them with a heightened capacity for detecting and responding to gas exposure [55]. This advantage translates to faster response times and greater sensitivity. Nevertheless, it is worth noting that a precise comparison between the two is contingent on factors such as particle size, morphology, distribution, and the specific gas under consideration [56]. The response of the optimized sensor is compared with those of previously reported ones in Table 2, demonstrating that the as-fabricated gas sensor exhibits superior acetone sensing performances.
Table 2.
Comparison of NiO-based gas sensors.
In essence, the superior surface-area-to-volume ratio inherent to nanodispersed nickel oxide particles engenders enhanced surface reactivity and heightened sensitivity to fluctuations in gas concentration [61]. This heightened sensitivity emerges from the greater abundance of active sites available for interaction with gas molecules [62]. Additionally, the diminutive size of nanodispersed nickel oxide particles accelerates the diffusion of gas molecules towards these active sites, thereby contributing to swifter response times.
Conversely, microdispersed nickel oxide particles typically exhibit smaller surface areas, resulting in lower sensitivity to variations in gas concentration. Nevertheless, their utility in gas detection remains valuable due to factors such as cost-effectiveness, stability, and ease of synthesis when compared to their nanodispersed counterparts.
It is imperative to acknowledge that the gas sensing characteristics of microdispersed and nanodispersed nickel oxide particles can exhibit substantial variations contingent upon numerous factors, including particle size, morphology, crystal structure, surface chemistry, and the presence of impurities or surface defects. Consequently, a comprehensive analysis of the gas-sensitive attributes of both forms of nickel oxide is requisite for a definitive comparison.
According to the operating principle of p-type semiconductors such as NiO, oxygen molecules adsorbed onto the surface capture electrons from the semiconductor, transforming into ionic species in the presence of air [63]. This transformation results in an augmentation of basic carriers, thereby yielding high sensor current values in ambient air. However, gaseous acetone, being a reducing gas, initiates a reaction between the reducing gas and oxygen ions on the NiO surface when exposed to the sensor. Based on our findings, the response and recovery times for NiO amount to 40.6 s and 630.2 s, respectively.
3.3. Characterization of Obtained C/TiO2 Composite Fibers
Composite fibers were synthesized through the incorporation of titanium oxide nanoparticles into a matrix comprising polyacrylonitrile and coal tar pitch. The morphological characteristics of these fibers after stabilization and carbonization were studied using SEM (Figure 7). SEM images demonstrate the preservation of the fibrous structure, which is emphasized with the presence of crystalline inclusions of metallic nanoparticles adhering to the fiber face, as well as the formation of nanoparticle agglomerates.

Figure 7.
SEM images of composite fibers PAN/CTP/TiO2 after stabilization and carbonization.
Figure 8 shows the change in the concentration of methylene blue during the adsorption process in the absence of light. Here, C0 is the initial concentration of the dye, while Ct is the remaining concentration of MB at that time.
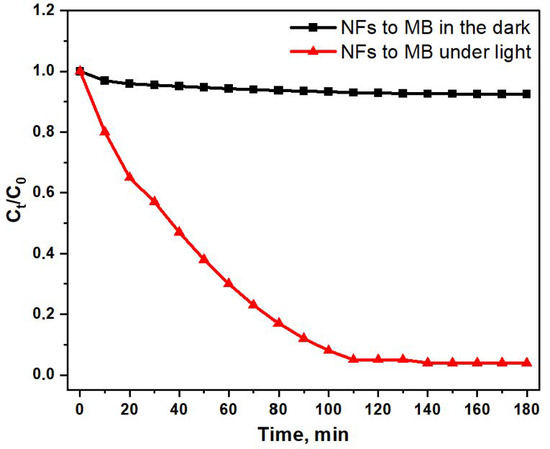
Figure 8.
The adsorption profile of C/TiO2 to MB in the dark and under light.
The degradation efficiency D% is obtained with the following equation:
where C0 is the concentration of MB. The final degradation efficiency is obtained by taking the Ct value when t is 180 min. It is seen that the degradation efficiency is about 96.13 ± 0.05%, which is the highest efficiency.
D % = C0 − Ct/C0 × 100%,
Table 3 provides a comparison of different photocatalysts in their ability to degrade specific model dye pollutants, along with the degradation rates and irradiation times. The last entry is for the current study.
Table 3.
Comparison of TiO2-based photocatalysts.
The obtained results are of practical interest for the removal of organic dyes using photocatalytic decomposition, as demonstrated with the example of methylene blue decomposition. The results indicate that C/TiO2 composite fibers were obtained after stabilization and carbonization of PAN/CTP/TiO2 fibers. These fibers after high-temperature treatment and removal of all volatile components possess both chemical stability and mechanical strength [68].
The structure of the resulting fibers resembles a “beads-on-a-string” [69] configuration, where the “beads” are crystalline TiO2 inclusions interconnected with carbon fibers. This structure provides access to photocatalytically active TiO2 for the degradation of dye molecules such as MB. Since the mat composed of C/TiO2 fibers can be approximated as a permeable membrane due to the voids between the fibers, even TiO2 particles in the deeper layers of the electrospun mat remain active [70]. These results are promising for the application of composite fibers in photocatalytic processes, especially in areas such as wastewater treatment and environmental protection.
3.4. Characterization of C/Fe3O4 Composite Fibers
The examination of magnetite nanoparticles involved a comprehensive investigation utilizing XRD and TEM. In Figure 9, the TEM image with the particle size distribution is presented based on the TEM analysis of the magnetite particles synthesized via chemical deposition. Predominantly, the particles fell within the size range of 6 to 16 nm, with an average particle diameter measuring approximately 12.22 nm.
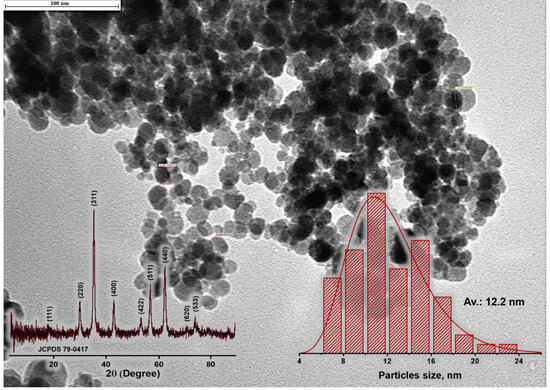
Figure 9.
TEM image (with particle size distribution and XRD patterns) of magnetite nanoparticles.
The X-ray pattern of the magnetite nanoparticles, as illustrated in Figure 9, reveals a distinctive pattern marked by nine well-defined diffraction peaks occurring at 2θ angles of 18.5, 30.1, 35.5, 43.2, 53.6, 57.1, 62.7, 71.3, and 74.2 degrees. These angles correspond precisely to the crystallographic planes of the magnetite phase, specifically identified as (111), (220), (311), (400), (422), (511), (440), (620), and (533), respectively (JCPDS card 03-065-3107).
The average particle size, symbolized as ‘t’, was calculated employing the Scherrer equation, t = λ/(βcos θ). In our analysis, the size of the synthesized magnetite particles was conclusively determined to be 10.4 nm, a result that consistently concurs with the data obtained from TEM imaging.
The morphology of the obtained fibres was examined by SEM (Figure 10). The elemental analysis of electrospun PAN/CTP/Fe3O4 fibers yielded the following compositional data:

Figure 10.
SEM images (with EDAX-analysis) of electrospun, stabilized, and carbonized PAN/CTP/Fe3O4 composite fibers.
In the electrospun state, carbon (C) constituted 65.95 wt.%, nitrogen (N) comprised 17.49 wt.%, oxygen (O) constituted 7.23 wt.%, and iron (Fe) accounted for 9.33 wt.%.
Post-stabilization, the composition exhibited slight variations, with carbon at 65.86 wt.%, nitrogen at 17.63 wt.%, oxygen at 6.96 wt.%, and iron at 9.55 wt.%.
Following carbonization, a pronounced transformation was observed, with carbon content reducing substantially to 41.50 wt.%, nitrogen diminishing to 3.01 wt.%, oxygen increasing to 15.25 wt.%, and iron markedly escalating to 40.06 wt.%.
Changes in the elemental composition of PAN/CTP/Fe3O4 fibers during the stabilization and carbonization processes can be explained with the main processes occurring at each stage. Initially, the primary constituents of the fibers were carbon, nitrogen, oxygen, and iron, reflecting the composition of the starting materials. During the stabilization stage, there was a loss of carbon and slight changes in nitrogen content, while the iron content remained constant. Subsequently, during the carbonization process, there was a decrease in carbon and nitrogen content, and the increased presence of iron was attributed to its concentration in the remaining material. In general, the changes in the content and composition of C/Fe3O4 fibers at different stages are related to the chemical and structural transformations that occur during processing. The processes of stabilization and carbonization lead to the removal of organic components and their transformation into carbon structures [71], which explains the variations in the content of carbon, nitrogen, oxygen, and iron at each stage.
In summation, the variations in mass content and elemental composition observed at distinct phases of C/Fe3O4 fiber production are fundamentally a consequence of the intricate chemical and structural metamorphoses occurring during the heating and processing of the fibers. The stabilization and carbonization processes are marked by the removal of organic components and the conversion of precursor fibers into carbon-enriched structures, thereby accounting for the marked fluctuations in carbon, nitrogen, oxygen, and iron content observed at each juncture.
3.5. Characterization of C/AC Composite Fibers
3.5.1. Characterization of Activated Carbon Obtained from Rice Husk
Composite fibers based on polyacrylonitrile and coal tar pitches with the addition of activated carbon particles synthesized from rice husk, a vegetable waste, were obtained. As can be seen from the results of the SEM analysis (Figure 11), the sample has a loose-layered structure with a texture of pores represented by interconnected cavities formed because of the release of volatile organic substances.

Figure 11.
SEM image of activated carbon.
Activated carbon due to its large surface area and porous architecture, providing exceptional adsorption, adsorbs heavy metal ions, promoting their removal from aqueous solutions [21]. This is facilitated with numerous active sites within activated carbon that bind heavy metal ions through mechanisms such as ion exchange and chemical adsorption [22]. Consequently, using activated carbon declines heavy metal ion concentration in water, promoting water safety and environmental preservation. Furthermore, the adoption of activated carbon in removing heavy metal ions can enhance wastewater quality and prevent soil and groundwater contamination, making it vital for water treatment and environmental protection.
3.5.2. Characterization of PAN/CTP/AC Fibers
In an analytical context, examination of SEM images, depicted in Figure 12, regarding composite fibers based on PAN/CTP/AC, revealed an absence of a methodical orientation axis during spinning. Rather, these fibers displayed a random and disorganized arrangement. After undergoing thermal treatments that included both stabilization and carbonization processes, a notable observation was made: the fibrous structure was preserved without any evident defects or dislocations. This indicates that the fibers maintained their resilience and structural integrity throughout the heat treatment regimen. Upon a thorough examination of micrographs at varying magnifications, a consistent dispersion of the fibers was observed. The uniform distribution remained consistent across various magnifications, demonstrating homogeneous fiber morphology throughout the range under examination. Furthermore, the analysis of fiber shape confirmed the uniformity and homogeneity of the fibers.

Figure 12.
SEM images of PAN/CTP/AC fibers after stabilization and carbonization.
There is a reduction in fiber diameter after carbonization owing to different factors. PAN undergoes thermal decomposition during carbonization, leading to the elimination of non-carbonaceous components in the form of volatile gases like water vapor, carbon dioxide, and ammonia [72]. This procedure enhances carbon content within the fibers. As the atomic radius of carbon atoms is smaller compared to heteroatoms present in PAN [73], the carbon structure undergoes rearrangement, leading to increased order, shrinkage, and densification, ultimately decreasing their diameter. The precise and consistent fiber measurements, with an average diameter of 0.5 µm, were demonstrated.
3.5.3. Investigation of Metal Ion Sorption Properties
Composite fibers with activated carbon are used in water purification to eliminate heavy metal ions [21,22,74,75,76,77,78,79,80,81]. The collaboration of the fiber matrix in the composite material and the large surface area of the activated carbon enhances the removal of heavy metal ions from water due to its sorption properties. The fibrous matrix increases the contact area of water with the sorbent, while the activated carbon particles act as potent sorption centers for heavy metal ions. Empirical research has confirmed that these fibers possess a high sorption capacity towards heavy metal ions like lead [82], cadmium [83], and mercury [83,84], attributed to the activated carbon’s large surface area and porous structure. A comprehensive analysis was conducted on the sorption properties of the composite fibers obtained (refer to Figure 13) during the adsorption process of manganese (II) ions from water at various time intervals. This study will present insights into the kinetics and efficiency of adsorbing manganese (II) ions through composite fibers.
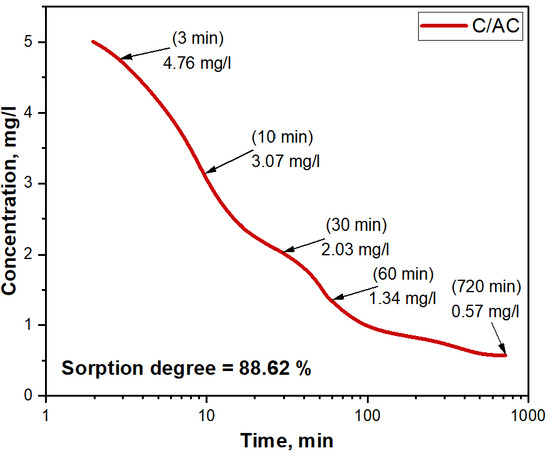
Figure 13.
Sorption of manganese (II) ions of ISO 7875-2000 in aqueous solution using C/AC composite fibers at different times.
Table 4 presents a comparative analysis of diverse carbon-based sorbents for their efficiency in eliminating manganese from aqueous solutions. The analysis includes data on the percentage of manganese removal, initial concentration, and operating temperature. The final entry pertains to the current study.
Table 4.
Comparison of carbon-based sorbents.
4. Conclusions
The research results encompass the synthesis and testing of composite electrospun fibers using coal tar pitch and various nanomaterial additives for eco-bio-applications. The thermal treatment of coal tar has unveiled its environmental potential, as high temperatures remove volatile components and sulfur, rendering it suitable for fiber electrospinning and subsequent carbonization. The successful application of CTP as an additive to a basic PAN solution is demonstrated through the synthesis of composite fibers incorporating CTP and nanomaterials like nickel oxide (NiO), titanium dioxide (TiO2), and activated carbon (AC). NiO nanoparticles were obtained through the solution combustion method, while activated carbon was synthesized from agricultural waste, specifically rice husks. The C/NiO fibers exhibited good sensitivity to 100 ppm acetone at 220 °C with a response time of 40.6 s. The C/TiO2 fibers, resembling a “beads-on-a-string” structure, demonstrated a high efficiency of 96.13% in the decomposition of methylene blue. Additionally, the C/AC fibers exhibited good adsorption characteristics, as evidenced with their efficient sorption of manganese (II) ions in aqueous solutions, with an efficiency of 88.62%. This research showcases how a combination of various methods, approaches, and materials, including waste materials, can lead to the production of composite materials with a broad range of applications. Beyond advancing the field of composite materials science, this work paves the way for sustainable solutions to pressing eco-bio-challenges. The developed composite fibers exemplify the potential of environmentally responsible materials and technologies to contribute to a greener and more sustainable future.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by B.K., G.S., A.I. (Aigerim Imash), A.I. (Akram Ilyanov) and Z.M. The first draft of the manuscript was written by B.K. and all authors commented on previous versions of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Ministry of Science and Higher Education of the Republic of Kazakhstan, grant number: AP09259842.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ahmad, F.; Saeed, Q.; Shah, S.M.U.; Gondal, M.A.; Mumtaz, S. Environmental Sustainability: Challenges and Approaches. In Natural Resources Conservation and Advances for Sustainability; Elsevier: Amsterdam, The Netherlands, 2022; pp. 243–270. [Google Scholar] [CrossRef]
- Aithal, S.; Aithal, P.S. Green Nanotechnology Innovations to Realize UN Sustainable Development Goals 2030. Int. J. Appl. Eng. Manag. Lett. 2021, 5, 96–105. [Google Scholar] [CrossRef]
- Frenot, A.; Chronakis, I.S. Polymer Nanofibers Assembled by Electrospinning. Curr. Opin. Colloid Interface Sci. 2003, 8, 64–75. [Google Scholar] [CrossRef]
- Farshchi, R.; Ramsteiner, M. Spin Injection from Heusler Alloys into Semiconductors: A Materials Perspective. J. Appl. Phys. 2013, 113, 191101. [Google Scholar] [CrossRef]
- Thejas Prasannakumar, A.; Mohan, R.R.; Varma, S.J. Progress in Conducting Polymer-Based Electrospun Fibers for Supercapacitor Applications: A Review. ChemistrySelect 2023, 8, e202203564. [Google Scholar] [CrossRef]
- Langwald, S.V.; Ehrmann, A.; Sabantina, L. Measuring Physical Properties of Electrospun Nanofiber Mats for Different Bio-medical Applications. Membranes 2023, 13, 488. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Liu, Y.; Bai, Y.; Shi, H.; Zhou, W.; Chen, Y.; Liu, Y.; Yu, D.-G. Recent Combinations of Electrospinning with Photocatalytic Technology for Treating Polluted Water. Catalysts 2023, 13, 758. [Google Scholar] [CrossRef]
- Wang, M.-L.; Yu, D.-G.; Bligh, S.W.A. Progress in Preparing Electrospun Janus Fibers and Their Applications. Appl. Mater. Today 2023, 31, 101766. [Google Scholar] [CrossRef]
- Wang, P.; Lv, H.; Cao, X.; Liu, Y.; Yu, D.-G. Recent Progress of the Preparation and Application of Electrospun Porous Nanofibers. Polymers 2023, 15, 921. [Google Scholar] [CrossRef]
- Zoppas, F.M.; Beltrame, T.F.; Sosa, F.A.; Bernardes, A.M.; Miró, E.; Marchesini, F.A. Superficial properties of activated carbon fiber catalysts produced by green synthesis and their application in water purification. Environ. Sci. Pollut. Res. 2020, 27, 40405–40420. [Google Scholar] [CrossRef]
- Ji, W.; Wang, X.; Ding, T.; Chakir, S.; Xu, Y.; Huang, X.; Wang, H. Electrospinning preparation of nylon-6@UiO-66-NH2 fiber membrane for selective adsorption enhanced photocatalysis reduction of Cr(VI) in water. Chem. Eng. J. 2023, 451, 138973. [Google Scholar] [CrossRef]
- Mamah, S.C.; Goh, P.S.; Ismail, A.F.; Suzaimi, N.D.; Yogarathinam, L.T.; Raji, Y.O.; El-badawy, T.H. Recent development in modification of polysulfone membrane for water treatment application. J. Water Process Eng. 2021, 40, 101835. [Google Scholar] [CrossRef]
- Rana, A.; Sudhaik, A.; Raizada, P.; Khan, A.A.P.; Van Le, Q.; Singh, A.; Selvasembian, R.; Nadda, A.; Singh, P. An overview on cellulose-supported semiconductor photocatalysts for water purification. Nanotechnol. Environ. Eng. 2021, 6, 40. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, P.; Yao, Y.; Yamauchi, Y.; Yan, X.; Li, J.; Na, J. In-situ fabrication of nanoarchitectured MOF filter for water purification. J. Hazard. Mater. 2020, 392, 122164. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Gao, Y.; Wang, W.; Feng, H.; Chen, K.; Xiao, C. Green preparation of PVDF hollow fiber membranes with multiple pore structure via melt spinning method for oil/water separation. J. Environ. Chem. Eng. 2022, 10, 108337. [Google Scholar] [CrossRef]
- Wang, A.; Li, X.; Hou, T.; Lu, Y.; Zhou, J.; Zhang, X.; Yang, B. A tree-grapes-like PTFE fibrous membrane with superhydrophobic and durable performance for oil/water separation. Sep. Purif. Technol. 2021, 275, 119165. [Google Scholar] [CrossRef]
- Ma, W.; Li, Y.; Gao, S.; Cui, J.; Qu, Q.; Wang, Y.; Huang, C.; Fu, G. Self-healing and superwettable nanofibrous membranes with excellent stability toward multifunctional applications in water purification. ACS Appl. Mater. Interfaces 2020, 12, 23644–23654. [Google Scholar] [CrossRef]
- Nthunya, L.N.; Gutierrez, L.; Derese, S.; Nxumalo, E.N.; Verliefde, A.R.; Mamba, B.B.; Mhlanga, S.D. A review of nanoparticle-enhanced membrane distillation membranes: Membrane synthesis and applications in water treatment. J. Chem. Technol. Biotechnol. 2019, 94, 2757–2771. [Google Scholar] [CrossRef]
- Sagitha, P.; Reshmi, C.R.; Manaf, O.; Sundaran, S.P.; Juraij, K.; Sujith, A. Development of nanocomposite membranes by electrospun nanofibrous materials. In Nanocomposite Membranes for Water and Gas Separation; Elsevier: Amsterdam, The Netherlands, 2020; pp. 199–218. [Google Scholar] [CrossRef]
- Uddin, Z.; Ahmad, F.; Ullan, T.; Nawab, Y.; Ahmad, S.; Azam, F.; Rasheed, A.; Zafar, M.S. Recent trends in water purification using electrospun nanofibrous membranes. Int. J. Environ. Sci. Technol. 2022, 19, 9149–9176. [Google Scholar] [CrossRef]
- Shahrokhi-Shahraki, R.; Benally, C.; El-Din, M.G.; Park, J. High efficiency removal of heavy metals using tire-derived activated carbon vs commercial activated carbon: Insights into the adsorption mechanisms. Chemosphere 2021, 264, 128455. [Google Scholar] [CrossRef]
- Yuan, Y.; An, Z.; Zhang, R.; Wei, X.; Lai, B. Efficiencies and mechanisms of heavy metals adsorption on waste leather-derived high-nitrogen activated carbon. J. Clean. Prod. 2021, 293, 126215. [Google Scholar] [CrossRef]
- Thambiliyagodage, C. Efficient Photocatalysis of Carbon Coupled TiO2 to Degrade Pollutants in Wastewater—A Review. Environ. Nanotechnol. Monit. Manag. 2022, 18, 100737. [Google Scholar] [CrossRef]
- Yamakata, A.; Vequizo, J.J.M. Curious Behaviors of Photogenerated Electrons and Holes at the Defects on Anatase, Rutile, and Brookite TiO2 Powders: A Review. J. Photochem. Photobiol. C Photochem. Rev. 2019, 40, 234–243. [Google Scholar] [CrossRef]
- Qi, W.; Yang, Y.; Du, J.; Yang, J.; Guo, L.; Zhao, L. Highly Photocatalytic Electrospun Zr/Ag Co-Doped Titanium Dioxide Nanofibers for Degradation of Dye. J. Colloid Interface Sci. 2021, 603, 594–603. [Google Scholar] [CrossRef]
- Park, M.; Ko, Y.T.; Ji, M.; Cho, J.S.; Wang, D.H.; Lee, Y.-I. Facile Self-Assembly-Based Fabrication of a Polyvinylidene Fluoride Nanofiber Membrane with Immobilized Titanium Dioxide Nanoparticles for Dye Wastewater Treatment. J. Clean. Prod. 2022, 378, 134506. [Google Scholar] [CrossRef]
- Chen, Y.; Li, A.; Fu, X.; Peng, Z. Electrospinning-Based (N,F)-Co-Doped TiO2-δ Nanofibers: An Excellent Photocatalyst for Degrading Organic Dyes and Heavy Metal Ions under Visible Light. Mater. Chem. Phys. 2022, 291, 126672. [Google Scholar] [CrossRef]
- Cen, L.; Tang, T.; Yu, F.; Wu, H.; Li, C.; Zhu, H.; Guo, Y. Fabrication of ZIF-8/TiO2 Electrospinning Nanofibers for Synergistic Photodegradation in Dyeing Wastewater. J. Ind. Eng. Chem. 2023, 126, 537–545. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, G.; Wang, Q.; Meng, F.; Jia, H.; Jiang, W.; Ji, Q. Hybrid Nanoarchitectonics of TiO2/Aramid Nanofiber Membranes with Softness and Durability for Photocatalytic Dye Degradation. Chin. Chem. Lett. 2023, 109193. [Google Scholar] [CrossRef]
- Sun, F.; Qi, H.; Xie, Y.; Ma, Q.; He, W.; Xu, D.; Wang, G.; Yu, W.; Wang, T.; Dong, X. Flexible Self-Supporting Bifunctional [TiO2/C]//[Bi2WO6/C] Carbon-Based Janus Nanofiber Heterojunction Photocatalysts for Efficient Hydrogen Evolution and Degradation of Organic Pollutant. J. Alloys Compd. 2020, 830, 154673. [Google Scholar] [CrossRef]
- Singh, E.; Meyyappan, M.; Nalwa, H.S. Flexible Graphene-Based Wearable Gas and Chemical Sensors. ACS Appl. Mater. Interfaces 2017, 9, 34544–34586. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, Y.; Gopalan, S.; Luo, F.; Amreen, K.; Singh, R.K.; Goel, S.; Lin, Z.; Naidu, R. Review and Perspective: Gas Separation and Discrimination Technologies for Current Gas Sensors in Environmental Applications. ACS Sens. 2023, 8, 1373–1390. [Google Scholar] [CrossRef]
- Krishna, K.G.; Parne, S.; Pothukanuri, N.; Kathirvelu, V.; Gandi, S.; Joshi, D. Nanostructured Metal Oxide Semiconductor-Based Gas Sensors: A Comprehensive Review. Sens. Actuators Phys. 2022, 341, 113578. [Google Scholar] [CrossRef]
- Yoon, Y.; Truong, P.L.; Lee, D.; Ko, S.H. Metal-Oxide Nanomaterials Synthesis and Applications in Flexible and Wearable Sensors. ACS Nanosci. Au 2022, 2, 64–92. [Google Scholar] [CrossRef]
- Yang, J.; Han, W.; Jiang, B.; Wang, X.; Sun, Y.; Wang, W.; Lou, R.; Ci, H.; Zhang, H.; Lu, G. Electrospinning Derived NiO/NiFe 2O4 Fiber-in-Tube Composite for Fast Triethylamine Detection under Different Humidity. ACS Sens. 2022, 7, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zeng, W.; Li, Y. NiO-Based Gas Sensors for Ethanol Detection: Recent Progress. J. Sens. 2022, 2022, 1855493. [Google Scholar] [CrossRef]
- Rohman, Y.M.; Sukowati, R.; Priyanto, A.; Hapidin, D.A.; Edikresnha, D.; Khairurrijal, K. Quartz Crystal Microbalance Coated with Polyacrylonitrile/Nickel Nanofibers for High-Performance Methanol Gas Detection. ACS Omega 2023, 8, 13342–13351. [Google Scholar] [CrossRef]
- Kampara, R.K.; Sonia, T.; Balamurugan, D.; Jeyaprakash, B.G. Formaldehyde Vapour Sensing Property of Electrospun NiO Nanograins. Front. Mater. Sci. 2021, 15, 416–430. [Google Scholar] [CrossRef]
- Shoukat, N.; Anzar, S.; Asad, M.; Al-Sulami, A.I.; Khalid, H.; Choudhary, A.A.; Sherin, L.; Akhtar, N. Fabrication of CuO–NiO Wrapped Cellulose Acetate/Polyaniline Electrospun Nanofibers for Sensitive Monitoring of Bisphenol-A. ACS Sustain. Chem. Eng. 2023, 11, 4299–4307. [Google Scholar] [CrossRef]
- Wu, L.; Song, Y.; Xing, S.; Li, Y.; Xu, H.; Yang, Q.; Li, Y. Advances in Electrospun Nanofibrous Membrane Sensors for Ion Detection. RSC Adv. 2022, 12, 34866–34891. [Google Scholar] [CrossRef]
- Blachowicz, T.; Ehrmann, A. Most Recent Developments in Electrospun Magnetic Nanofibers: A Review. J. Eng. Fibers Fabr. 2020, 15, 155892501990084. [Google Scholar] [CrossRef]
- Gan, Y.; Yu, C.; Panahi, N.; Gan, J.; Cheng, W. Processing Iron Oxide Nanoparticle-Loaded Composite Carbon Fiber and the Photosensitivity Characterization. Fibers 2019, 7, 25. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Xu, L.; Tang, X.-P.; Sun, Z.Q. Effect of Fe3Os Nanoparticles on Magnetic Electrospun Nanofibers. J. Text. Inst. 2015, 106, 503–509. [Google Scholar] [CrossRef]
- Shi, S.; Xu, C.; Wang, X.; Xie, Y.; Wang, Y.; Dong, Q.; Zhu, L.; Zhang, G.; Xu, D. Electrospinning Fabrication of Flexible Fe3O4 Fibers by Sol-Gel Method with High Saturation Magnetization for Heavy Metal Adsorption. Mater. Des. 2020, 186, 108298. [Google Scholar] [CrossRef]
- Shi, S.; Xu, C.; Dong, Q.; Wang, Y.; Zhu, S.; Zhang, X.; Tak Chow, Y.; Wang, X.; Zhu, L.; Zhang, G.; et al. High Saturation Magnetization MnO2/PDA/Fe3O4 Fibers for Efficient Pb(II) Adsorption and Rapid Magnetic Separation. Appl. Surf. Sci. 2021, 541, 148379. [Google Scholar] [CrossRef]
- Teng, Y.; Li, Y.; Li, Y.; Song, Q. Preparation of Fe3O4/PVP Magnetic Nanofibers via in Situ Method with Electro-spinning. J. Phys. Conf. Ser. 2020, 1549, 032087. [Google Scholar] [CrossRef]
- Liu, J.; Chang, M.-J.; Du, H.-L. Facile Preparation of Cross-Linked Porous Poly(Vinyl Alcohol) Nanofibers by Electrospinning. Mater. Lett. 2016, 183, 318–321. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Zhang, B.; Sun, Z.; Li, Z.; Jiang, X.; Bai, X. Preparation of Fe3O4/PVA Nanofibers via Com-bining in-Situ Composite with Electrospinning. Mater. Lett. 2010, 64, 9–11. [Google Scholar] [CrossRef]
- Konwar, A.; Gogoi, A.; Chowdhury, D. Magnetic Alginate–Fe3O4 Hydrogel Fiber Capable of Ciprofloxacin Hydrochloride1161 Adsorption/Separation in Aqueous Solution. RSC Adv. 2015, 5, 81573–81582. [Google Scholar] [CrossRef]
- Mansurov, Z.; Smagulova, G.; Kaidar, B.; Imash, A.; Lesbayev, A. PAN—Composite Electrospun-Fibers Decorated with Magnetite Nanoparticles. Magnetochemistry 2022, 8, 160. [Google Scholar] [CrossRef]
- Prikhod’ko, N.G.; Smagulova, G.T.; Nazhipkyzy, M.; Rakhymzhan, N.B.; Temirgalieva, T.S.; Lesbaev, B.T.; Zakhidov, A.A.; Mansurov, Z.A. High-Efficiency Selective Solar Absorber from Nanostructured Carbonized Plant Raw Material. J. Eng. Phys. Thermophys. 2020, 93, 1020–1029. [Google Scholar] [CrossRef]
- Liang, L.; Huang, W.; Gao, F.; Hao, X.; Zhang, Z.; Zhang, Q.; Guan, G. Mild Catalytic Depolymerization of Low Rank Coals: A Novel Way to Increase Tar Yield. RSC Adv. 2015, 5, 2493–2503. [Google Scholar] [CrossRef]
- Karika, J.C. Characterization of Graphitization in Coal Tar and Petroleum Pitches. Master’s Thesis, Arizona State University, Tempe, AZ, USA, 1985. Available online: https://apps.dtic.mil/sti/citations/ADA166942 (accessed on 2 April 2023).
- Ali, B.; Tasirin, S.; Aminayi, P.; Yaakob, Z.; Ali, N.; Noori, W. Non-Supported Nickel-Based Coral Sponge-Like Porous Magnetic Alloys for Catalytic Production of Syngas and Carbon Bio-Nanofilaments via a Biogas De-composition Approach. Nanomaterials 2018, 8, 1053. [Google Scholar] [CrossRef] [PubMed]
- Zaitseva, N.V.; Zemlyanova, M.A.; Zvezdin, V.N.; Dovbysh, A.A.; Ulanova, T.S.; Smirnov, S.A.; Stepankov, M.S. Comparative Assessment of the Effects of Short-Term Inhalation Exposure to Nickel Oxide Nanoparticles and Microdispersed Nickel Oxide. Nanotechnol. Russ. 2016, 11, 671–677. [Google Scholar] [CrossRef]
- Mokoena, T.P.; Swart, H.C.; Motaung, D.E. A Review on Recent Progress of P-Type Nickel Oxide Based Gas Sensors: Future Perspectives. J. Alloys Compd. 2019, 805, 267–294. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Leng, D.; Li, G.; Zhang, Y.; Wang, W.; Liang, Q.; Chen, X.; Lu, H.; Gao, J. Electrospun NiO Nanofibers with Rh Decoration for Enhanced Acetone Sensing Performances. J. Mater. Sci. Mater. Electron. 2021, 32, 14102–14112. [Google Scholar] [CrossRef]
- Fan, X.; Xu, Y.; He, W. High Acetone Sensing Properties of In2O3 –NiO One-Dimensional Heterogeneous Nanofibers Based on Electrospinning. RSC Adv. 2021, 11, 11215–11223. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, J.; Zhou, T.; Tu, J.; Zhang, T. A Yolk-Double-Shelled Heterostructure-Based Sensor for Acetone Detecting Application. J. Colloid Interface Sci. 2019, 539, 490–496. [Google Scholar] [CrossRef]
- Yang, J.; Han, W.; Ma, J.; Wang, C.; Shimanoe, K.; Zhang, S.; Sun, Y.; Cheng, P.; Wang, Y.; Zhang, H.; et al. Sn Doping Effect on NiO Hollow Nanofibers Based Gas Sensors about the Humidity Dependence for Triethylamine Detection. Sens. Actuators B Chem. 2021, 340, 129971. [Google Scholar] [CrossRef]
- Soleimanpour, A.M.; Jayatissa, A.H.; Sumanasekera, G. Surface and Gas Sensing Properties of Nanocrystalline Nickel Oxide Thin Films. Appl. Surf. Sci. 2013, 276, 291–297. [Google Scholar] [CrossRef]
- Marikutsa, A.; Rumyantseva, M.; Konstantinova, E.A.; Gaskov, A. The Key Role of Active Sites in the Development of Selective Metal Oxide Sensor Materials. Sensors 2021, 21, 2554. [Google Scholar] [CrossRef]
- Nikolic, M.V.; Milovanovic, V.; Vasiljevic, Z.Z.; Stamenkovic, Z. Semiconductor Gas Sensors: Materials, Technology, Design, and Application. Sensors 2020, 20, 6694. [Google Scholar] [CrossRef]
- Mahadadalkar, M.A.; Park, N.; Yusuf, M.; Nagappan, S.; Nallal, M.; Park, K.H. Electrospun Fe Doped TiO2 Fiber Photocatalyst for Efficient Wastewater Treatment. Chemosphere 2023, 330, 138599. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, W.; Liu, X.; Tang, T.; Xiong, J. Fabrication of Titanium Dioxide/Carbon Fiber (TiO2/CF) Composites for Removal of Methylene Blue (MB) from Aqueous Solution with Enhanced Photocatalytic Activity. J. Chem. 2021, 2021, 9986158. [Google Scholar] [CrossRef]
- Cao, N.; Gu, M.; Gao, M.; Li, C.; Liu, K.; Zhao, X.; Feng, J.; Ren, Y.; Wei, T. A Three-Layer Photocatalyst Carbon Fibers/TiO2 Seed/TiO2 Nanorods with High Photocatalytic Degradation under Visible Light. Appl. Surf. Sci. 2020, 530, 147289. [Google Scholar] [CrossRef]
- Basit, M.A.; Raza, F.; Ali, G.; Parveen, A.; Khan, M.; Park, T.J. Nanoscale Modification of Carbon Fibers with CdS Quantum-Dot Sensitized TiO2: Photocatalytic and Photothermal Evaluation under Visible Irradiation. Mater. Sci. Semicond. Process. 2022, 142, 106485. [Google Scholar] [CrossRef]
- Santos, J.D.; Sato, T.P.; Lima, A.L.D.; Nogueira, A.S.; Quishida, C.C.C.; Borges, A.L.S. Titanium Dioxide and Polyethylmethacrylate Electrospun Nanofibers: Assessing the Technique Parameters and Morphological Characterization. Braz. Dent. Sci. 2019, 22, 70–78. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Xu, L.; Zheng, G.; Jiang, J.; Sun, D.; Li, W. Formation of Suspending Beads-on-a-String Structure in Electrohydrodynamic Printing Process. Mater. Des. 2021, 204, 109692. [Google Scholar] [CrossRef]
- Covaliu-Mierlă, C.I.; Matei, E.; Stoian, O.; Covaliu, L.; Constandache, A.-C.; Iovu, H.; Paraschiv, G. TiO2—Based Nanofibrous Membranes for Environmental Protection. Membranes 2022, 12, 236. [Google Scholar] [CrossRef]
- Maruccia, E.; Ferrari, S.; Bartoli, M.; Lucherini, L.; Meligrana, G.; Pirri, C.F.; Saracco, G.; Gerbaldi, C. Effect of Thermal Stabilization on PAN-Derived Electrospun Carbon Nanofibers for CO2 Capture. Polymers 2021, 13, 4197. [Google Scholar] [CrossRef]
- Hassan, M.F.; Sabri, M.A.; Fazal, H.; Hafeez, A.; Shezad, N.; Hussain, M. Recent trends in activated carbon fibers production from various precursors and applications—A comparative review. J. Anal. Appl. Pyrolysis 2020, 145, 104715. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, Z.; Yu, H.; Zhang, X.; Liu, T.; Xia, M.; Zheng, R.; Shui, M.; Shu, J. Heteroatom-doped carbon-based materials for lithium and sodium ion batteries. Energy Storage Mater. 2020, 32, 65–90. [Google Scholar] [CrossRef]
- Martins, I.M.; Sampaio, A.G.; Lima, G.M.; Oliveira e Campos, M.A.; Rodgher, S.; Rodrigues-Siqueli, A.C.; Baldan, M.R.; Marcuzzo, J.S.; Koga-Ito, C.Y. Application of textile (PAN-based) activated carbon fibers decorated with silver nanoparticles in water treatment. Front. Environ. Sci. 2023, 10, 1100583. [Google Scholar] [CrossRef]
- Yang, K.; Pan, T.; Zhao, Q.; Chen, C.; Zhu, X.; Wang, P.; Chen, B. Dual-function ultrafiltration membrane constructed from pure activated carbon particles via facile nanostructure reconstruction for high-efficient water purification. Carbon 2020, 168, 254–263. [Google Scholar] [CrossRef]
- Abdullah, N.; Othman, F.E.C.; Yusof, N.; Matsuura, T.; Lau, W.J.; Jaafar, J.; Ismail, A.F.; Salleh, W.N.W.; Aziz, F. Preparation of nanocomposite activated carbon nanofiber/manganese oxide and its adsorptive performance toward leads (II) from aqueous solution. J. Water Process Eng. 2020, 37, 101430. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, X. Purification performance of modified polyacrylonitrile fiber–activated carbon fiber filter for heavy metal ions. Environ. Sci. Pollut. Res. 2023, 30, 23372–23385. [Google Scholar] [CrossRef]
- Yunus, Z.M.; Al-Gheethi, A.; Othman, N.; Hamdan, R.; Ruslan, N.N. Removal of heavy metals from mining effluents in tile and electroplating industries using honeydew peel activated carbon: A microstructure and techno-economic analysis. J. Clean. Prod. 2020, 251, 119738. [Google Scholar] [CrossRef]
- Nordin, N.A.; Abdul Rahman, N.; Abdullah, A.H. Effective removal of Pb(II) ions by electrospun PAN/Sago lignin-based activated carbon nanofibers. Molecules 2020, 25, 3081. [Google Scholar] [CrossRef]
- Deng, Z.; Sun, S.; Li, H.; Pan, D.; Patil, R.R.; Guo, Z.; Seok, I. Modification of coconut shell-based activated carbon and purification of wastewater. Adv. Compos. Hybrid Mater. 2021, 4, 65–73. [Google Scholar] [CrossRef]
- Sakamoto, T.; Amano, Y.; Machida, M. Phosphate Ion Adsorption Properties of PAN-Based Activated Carbon Fiber Prepared with Na2CO3 Activation. J. Water Chem. Technol. 2021, 43, 298–304. [Google Scholar] [CrossRef]
- Zuo, Q.; Zheng, H.; Zhang, P.; Zhang, Y. Functionalized activated carbon fibers by hydrogen peroxide and polydopamine for efficient trace lead removal from drinking water. Langmuir 2021, 38, 253–263. [Google Scholar] [CrossRef]
- Kim, D.W.; Wee, J.H.; Yang, C.M.; Yang, K.S. Efficient removals of Hg and Cd in aqueous solution through NaOH-modified activated carbon fiber. Chem. Eng. J. 2020, 392, 123768. [Google Scholar] [CrossRef]
- Chen, B.C.; Tsai, C.Y.; Pan, S.Y.; Chen, Y.T.; Hsi, H.C. Sustainable recovery of gaseous mercury by adsorption and electrothermal desorption using activated carbon fiber cloth. Environ. Sci. Technol. 2020, 54, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Adekola, F.A.; Hodonou, D.S.S.; Adegoke, H.I. Thermodynamic and Kinetic Studies of Biosorption of Iron and Manganese from Aqueous Medium Using Rice Husk Ash. Appl. Water Sci. 2016, 6, 319–330. [Google Scholar] [CrossRef]
- Abdeen, Z.; Mohammad, S.G.; Mahmoud, M.S. Adsorption of Mn (II) Ion on Polyvinyl Alcohol/Chitosan Dry Blending from Aqueous Solution. Environ. Nanotechnol. Monit. Manag. 2015, 3, 1–9. [Google Scholar] [CrossRef]
- Tavlieva, M.P.; Genieva, S.D.; Georgieva, V.G.; Vlaev, L.T. Thermodynamics and Kinetics of the Removal of Manganese(II) Ions from Aqueous Solutions by White Rice Husk Ash. J. Mol. Liq. 2015, 211, 938–947. [Google Scholar] [CrossRef]
- Ali, A.; Saeed, K. Decontamination of Cr(VI) and Mn(II) from Aqueous Media by Untreated and Chemically Treated Banana Peel: A Comparative Study. Desalin. Water Treat. 2015, 53, 3586–3591. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).