1. Introduction
Sickle cell disease (SCD) is a genetic blood disorder caused by the inheritance of two abnormal beta-globin genes, at least one of which carries the sickle mutation (HbS). Different genotypes include HbSS or HbS/β-thalassemia [
1,
2]. SCD affects millions globally; based on the ministry of health in Saudi Arabia, 4.2% of the population has the trait, and 0.26% has SCD, with the most significant prevalence in the Eastern province [
3,
4].
In patients with SCD, this hemoglobin (Hb) gene defect leads to serious and life-threatening consequences such as inflammation, hemolytic anemia, splenic dysfunction resulting in impaired immunity to encapsulated organisms, vascular occlusion, and pain, all of which have an adverse impact on the patient’s health-related quality of life (HRQOL). Stroke, skin ulcers, priapism, acute and chronic organ damage, and a shortened life expectancy are possible secondary complications [
5,
6].
Pain is the most prevalent consequence of SCD and the most common reason for emergency room visits among SCD patients [
5]. They continue to suffer from multiple painful vaso-occlusive crises (VOCs), resulting in hospitalizations and end-organ damage, despite using blood transfusions, hydroxyurea, and opioids as cornerstones in managing SCD-related complications in children and adults [
5,
6]. The complex and poorly understood pathophysiology of SCD poses challenges to managing patients, both acutely and chronically [
6]. New targeted medications have been developed to address SCD in response to emerging pathophysiological insights [
7]. Over thirty pharmaceutical agents are currently under investigation for treating and preventing SCD-related complications, three of which have recently received FDA approval targeting vaso-occlusion and HbS polymerization: L-glutamine, crizanlizumab, and voxelotor [
7,
8]. However, recent regulatory updates have affected their availability: the conditional marketing authorization for crizanlizumab was revoked in August 2023 [
9], and voxelotor was withdrawn from the market by Pfizer in 2024 due to safety concerns [
10].
In SCD patients, oxidative stress plays a central role in the pathophysiology of hemolysis, inflammation, and vaso-occlusion. The Nicotinamide Adenine Dinucleotide (NAD) redox potential serves as a key indicator of oxidative stress by reflecting the balance between oxidized and reduced NAD molecules. Glutamine, an L-α-amino acid, is one of the most abundant amino acids in the body. Due to the accelerated red blood cell (RBC) turnover caused by chronic hemolysis in SCD, the demand for glutamine increases, making it a conditionally essential amino acid in this context. The proposed therapeutic rationale for L-glutamine supplementation in SCD lies in its antioxidant properties. L-glutamine supports the synthesis of antioxidants such as reduced glutathione, NAD(H), NADP(H), and nitric oxide, thereby enhancing redox balance and reducing oxidative damage [
11,
12]. Clinical and preclinical evidence indicates that L-glutamine increases NAD redox potential and NADH levels in sickle RBCs, potentially decreasing oxidative susceptibility and contributing to clinical improvements [
13,
14,
15,
16]. In a phase 3 clinical trial, L-glutamine was shown to reduce the frequency of vaso-occlusive crises by 25%, hospitalizations by 30%, and acute chest syndrome episodes over 48 weeks in both pediatric and adult SCD patients, with or without hydroxyurea [
17]. Consequently, L-glutamine (Endari
®) became the first therapy approved for SCD treatment since hydroxyurea in 1998 [
18].
Current data regarding the use and effectiveness of L-glutamine for sickle cell disease (SCD) in the Saudi population remain scarce. This knowledge gap limits evidence-based treatment strategies and optimal outcomes for these patients. Despite national screening programs, substantial gaps remain in access to disease-modifying therapies, comprehensive care centers, and patient education. Recent local studies also highlight the limited availability of newer agents such as L-glutamine, and regional disparities in hydroxyurea use remain a concern [
19]. Findings from the Real-World Assessment Survey for SCD in Saudi (ROARS) revealed significant gaps in the management of sickle cell disease by non-specialist healthcare providers, with limited use of newer therapies such as L-glutamine and marked variability in treatment approaches across regions [
20].
Moreover, our hospital requested L-Glutamine for 22 patients on a non-formulary basis until November 2022. There was a huge cost impact on our organization’s stretched budget. In addition to that, L-Glutamine has not yet been approved by the Saudi Food and Drug Authority (SFDA), and the procurement of unregistered medication is a significant challenge in Saudi Arabia. Staying up to date with drug regulations and compliance requirements is essential, as reported by a couple of groups of formulary management experts in Saudi Arabia [
21,
22]. This prompted our Pharmacy and Therapeutic (P&T) Committee to initiate a real-world evidence study on the effectiveness of L-Glutamine therapy in SCD patients to support their formulary decision of addition vs. rejection of the drug for its use in these patient population. To address this need, our study aimed to evaluate both the effectiveness and appropriateness of L-glutamine therapy in reducing SCD-related complications among adult and pediatric patients treated at King Abdulaziz Medical City, Jeddah.
2. Materials and Methods
We conducted a retrospective, observational, single-center study at King Abdulaziz Medical City in Jeddah, Saudi Arabia, between June 2019 and June 2023. This facility is one of the largest tertiary care centers, where a significant number of pediatric and adult patients with SCD are referred to the hematology section for disease management. Our study included those who had SCD and were actively receiving hydroxyurea prior to initiating L-Glutamine (Endari®).
L-Glutamine was initiated for patients who were at least 5 years old and had documented at least two crises in the previous year, with no upper limit on the number of crises, despite the use of hydroxyurea. We used the standard dosing as per the FDA label [<30 kg: 5 g (1 packet) twice daily (total dose 10 g/day), 30 to 65 kg: 10 g (2 packets) twice daily (total dose 20 g/day), >65 kg: 15 g (3 packets) twice daily (total dose 30 g/day)] [
23]. All patients were regularly followed up in hematology clinics with a follow-up period of 24 and 48 weeks. Study participants who were lost to follow-up after treatment initiation, as determined from their medical records, were excluded from the analysis.
The primary endpoint was to determine the frequency of VOCs in sickle cell disease patients through week 48 after L-glutamine initiation and compare it to historical control for 48 weeks prior to L-glutamine initiation.
Secondary endpoints included identifying the frequency and length of hospital stay due to VOCs and evaluating the percentage of laboratory parameter changes (hemoglobin level, hematocrit level, reticulocyte count, and lactate dehydrogenase (LDH) level) from baseline through weeks 24 and 48 and the cost impact of the use of L-glutamine.
For the purpose of this study, a data collection tool was developed after a thorough review of the symptoms acquired from each patient’s medical record at baseline and at weeks 24 and 48. Data on patient demographics (age, gender, weight), diagnosis using WHO ICD version 10, hydroxyurea use, laboratory parameters (hemoglobin levels, hematocrit, reticulocyte count, LDH levels), type of VOC and its frequency, length of hospital stay, and treatment discontinuation were collected. Clinical parameters, such as VOC type, VOC frequency, and duration of hospital stay, were collected 48 weeks before therapy commencement (referred to as historical control).
VOCs were defined according to Niihara et al. as acute pain episodes for no apparent cause other than a vaso-occlusive event involving a medical facility visit and being treated with oral or parenteral narcotic agents or a nonsteroidal anti-inflammatory drug. In addition, the occurrence of chest syndrome (chest-wall pain in association with findings of a new pulmonary infiltrate on chest X-ray films and fever), priapism, and splenic sequestration are considered sickle cell crises even if the symptoms are not painful enough to require narcotics [
17]. Secondary endpoints included determining the frequency of VOCs in sickle cell disease patients through week 24 after L-glutamine initiation, identifying the length of hospital stay due to VOCs after starting L-glutamine through week 48, and evaluating the percentage of laboratory parameter changes (hemoglobin level, hematocrit level, reticulocyte count, and LDH level) from baseline through weeks 24 and 48. For the cost impact, prices of L-glutamine were obtained from the National Unified Procurement Company (NUPCO) in the public sector.
Descriptive statistics were calculated to summarize the demographic and clinical characteristics of the study population, including percentages, the mean ± standard deviation (SD) for normally distributed data, and the median [interquartile range, IQR] for non-normally distributed data. To compare the effects of L-glutamine and historical control, a paired t-test was utilized. Two-tailed statistical significance was indicated by a p value of <0.05. The statistical program KNIME Analytics was used for all statistical analyses.
A post hoc power analysis was conducted for the primary outcome (VOC frequency at 48 weeks). Assuming a mean difference of 1 VOC event, an SD of 1.5, α = 0.05, and n = 11, the estimated power was approximately 38%.
3. Results
A total of 22 SCD patients were identified initially, and L-glutamine was requested for all 22 patients. However, after following the predefined inclusion criteria, only 15 patients were found to be eligible and were included in the final analysis (7 adults and 8 pediatric patients). The excluded patients comprised five individuals who had not initiated L-glutamine treatment at the time of data collection and two patients who had received the medication for less than 24 weeks.
Among the 15 patients included in the analysis, 11 completed the entire 48-week study duration. The reasons for incomplete follow-up varied, including loss of follow-up, unavailability of the medication, non-compliance, or insufficient information documented in their medical records. More than half of the patients were females (8, 53%), with a median age of 12 (IQR 7–20) years old and an average weight of 34.5 ± 16.6. All patients were on hydroxyurea before L-glutamine initiation and had reached their maximum tolerated dose, with a median of 24 (IQR 20–30) mg/kg (
Table 1). All of them continued using both hydroxyurea and L-glutamine together, except for one adult patient who was only on L-glutamine due to intolerance to hydroxyurea.
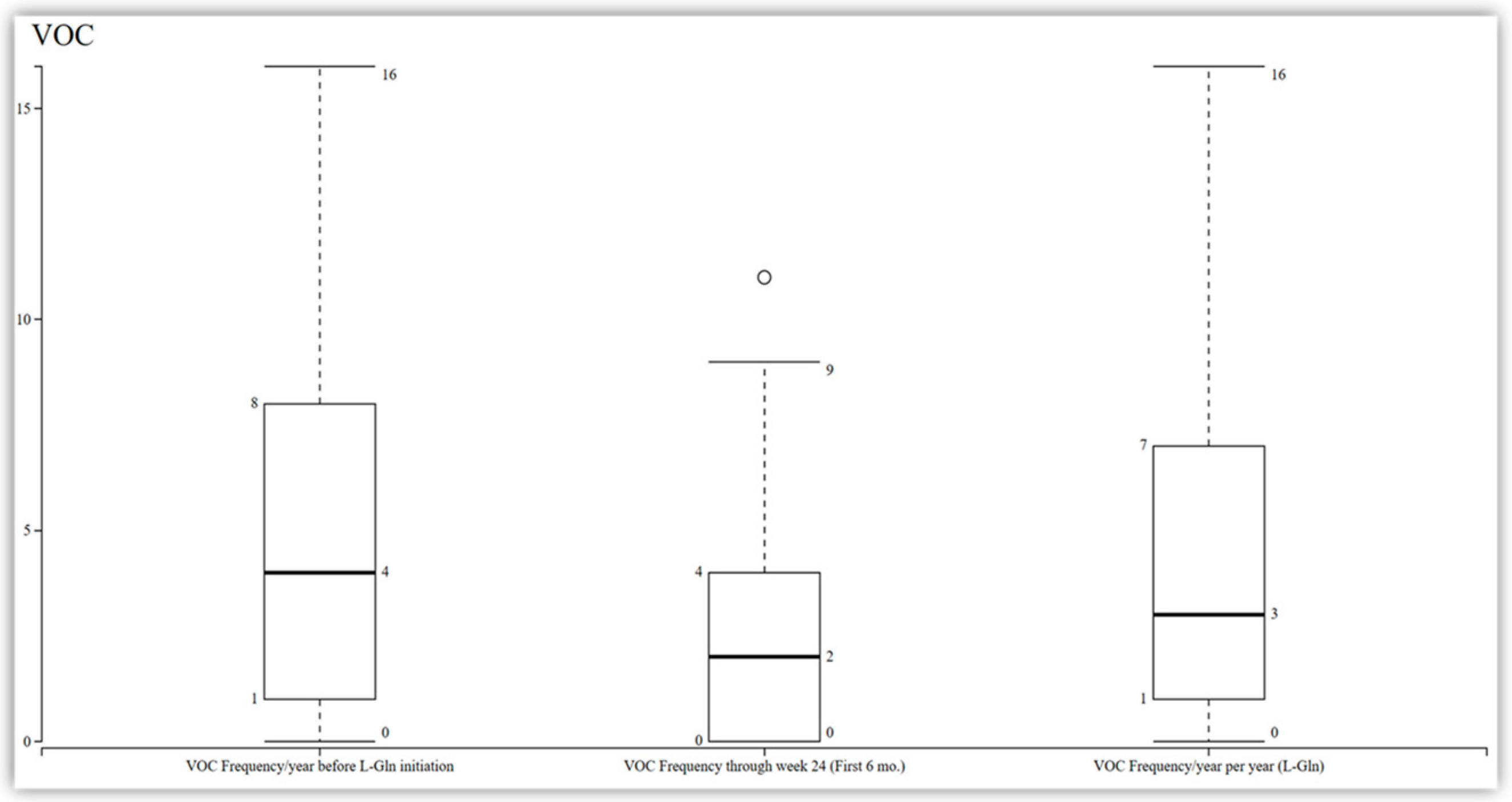
Among the 11 patients who completed the 48-week study, a reduction in the frequency of vaso-occlusive crises was observed compared to their baseline. However, this reduction did not reach statistical significance, with a median change from 4 to 3 (95% CI −1.37 to 0.65;
p = 0.44) at 48 weeks and a median change from 4 to 2 at 24 weeks (
Figure 1). Additionally, the analysis of the data revealed that there was no significant change in the length of stay among the patients. The median length of stay was observed to be seven days (IQR 0–29;
p = 0.72) (
Figure 2).
The laboratory parameters were assessed at weeks 24 and 48. The results indicate that there were no significant differences in these parameters compared to the baseline levels at both time points. For hemoglobin, the baseline median was 8.7 g/dL (IQR 8.4–9.8), with a 2.3% change at week 24 (IQR 8.4–9.8;
p = 0.84) and a 4.6% change at week 48 (IQR 8.8–10;
p = 0.49). However, noteworthy increases were observed in one specific laboratory parameter. At week 48, a substantial 61.9% rise in the reticulocyte count was observed, with a median of 278.5 (IQR 153–299;
p = 0.03) compared to baseline (
Table 2).
The prices of L-glutamine were obtained from NUPCO in the public sector. The unit price of L-glutamine 5 g/sachet is SAR 85.28 (22.74 USD). We used all 22 patients for the cost impact since L-glutamine was requested for all 22 patients on a non-formulary basis. The median dose used in pediatrics (n = 11) was 5 g per oral (PO) twice daily, and the median dose used in adults (n = 11) was 10 g PO twice daily. The cost of therapy for one pediatric patient per year was SAR 62,255 (16,601 USD), and the cost of therapy for one adult patient per year was SAR 124,509 (33,202 USD). The total estimated cost of therapy for 22 patients per year was SAR 2,054,399 (547,840 USD). This is not a cost-effectiveness modeling study; rather, it is just providing a cost impact of utilization of L-glutamine. It also anticipates cost savings or cost avoidance by rejecting the drug for its utilization in SCD patients in our center. Our P&T committee rejected the drug for this indication, and hence, cost savings or cost avoidance is important to mention here.
4. Discussion
Sickle cell disease is a prevalent condition with significant health implications for both adult and pediatric patients. Despite the high prevalence of SCD, there is a notable lack of studies investigating the efficacy and appropriateness of L-glutamine in reducing SCD-related complications. Hence, the aim of this study was to fill this gap by evaluating the efficacy and appropriateness of L-glutamine.
The results from our study, along with previous research on L-glutamine in SCD patients, consistently demonstrate a reduction in the frequency of VOCs. Although our study showed only a trend toward a reduction in VOC frequency that did not reach statistical significance, this finding aligns with the results from Niihara et al. (2018) and Elenga et al. (2022) [
17,
24], which observed similar trends. Notably, unlike those studies, our results did not achieve statistical significance. Niihara et al. conducted a phase 3 trial and showed a significant reduction in pain crises by 25% in SCD patients treated with L-glutamine. Similarly, Elenga et al.’s study on pediatric and adult SCD patients revealed significant improvements in clinical outcomes, including decreased pain crises, hospitalizations, days of hospitalization, blood transfusions, and acute chest syndrome events following L-glutamine therapy.
Importantly, the lack of statistical significance in our results may be attributed to the study’s limited statistical power. A post hoc power analysis revealed that the study had only 38% power to detect a one-event reduction in VOC frequency at 48 weeks, which is well below the conventional 80% threshold. This further underscores the exploratory nature of our findings and the need for caution in interpreting the absence of statistically significant results.
One other vital aspect to consider is the impact of L-glutamine on the length of hospital stays. We found that L-glutamine did not significantly affect the duration of hospital stays among patients receiving this treatment. These findings contrast with some previously published studies that have reported a reduction in hospitalizations and days of hospitalization with L-glutamine therapy in SCD patients. For example, Elenga et al. demonstrated a decrease in hospitalizations and days of hospitalization in SCD patients receiving L-glutamine [
24]. as Also, Niihara et al. reported a reduction in hospitalizations compared to the placebo group [
17].
In terms of laboratory results, most parameters did not show significant differences compared to baseline levels at both weeks 24 and 48. However, one laboratory parameter exhibited noteworthy change. Greater reticulocytosis in SCD patients emphasizes the disease’s hallmark of chronic peripheral hemolysis [
25]. The pathogenesis of SCD may be influenced by the distinct morphological and adhesive properties of sickle reticulocytes [
26]. Our study showed a significant 61.9% rise in the reticulocyte count at week 48 compared to baseline, which contradicts the results of other studies. This unexpected increase in the reticulocyte count may point to more hemolysis and not less. We found that two patients, one adult and one pediatric SCD patient, underwent splenectomy. These two patients had significant elevations in reticulocyte counts reaching 582 in the pediatric patient and 390 in the adult patient. Reticulocytosis is a common phenomenon observed after splenectomy which is not accompanied by significant changes in cell volume. Hence, reticulocytosis after splenectomy does not necessarily indicate a stress-induced erythropoietic stimulus [
27]. Moreover, we had three more patients with frequent VOCs whose reticulocyte count was significantly elevated, with the maximum retic count reaching 504, 450, and 426 in these three patients. These patients had not shown any response to L-glutamine. Moreover, 5 out of 11 patients with significantly elevated retic counts resulted in a 61% increase in retic count in the total study population, which is influenced by the small sample size.
The drug monograph of L-glutamine, along with the result of this study, was presented to the P&T committee on 28 February 2023. The P&T committee rejected the addition of L-glutamine to the formulary because of the lack of effectiveness and even recommended against non-formulary use of this drug in SCD patients. Hence, this study has a cost avoidance impact of around SAR 2,054,399 (547,840 USD) per year for 22 patients, and total cost savings over the 18 months have been SAR 3,081,599 (821,760 USD) since the P&T committee circulated minutes of the meeting in March 2023 stating the rejection of the drug. This cost avoidance is expected to be even more as our hospital did not request the drug for any new patient after the P&T made their decision. This paper was presented on the Saudi Commission for Health Specialists research day in 31 August 2023, and the data were shared publicly. This study has a significant impact on avoiding unnecessary costs associated with the use of this drug when it comes to formulary consideration of the drug nationally.
When comparing our findings to previously published data, it is crucial to acknowledge the limitations of our study. First, the retrospective design and the limited availability of complete data may affect our conclusions. Second, the relatively small sample size reduced the statistical power to detect significant differences, as confirmed by our post hoc analysis. Additionally, we were unable to directly assess the impact of disease severity and specific genetic polymorphisms on the efficacy of L-glutamine due to data constraints. Furthermore, we could not retrieve compliance data, as we did not have direct interactions with the patients. Finally, the reliance on a historical control group may introduce temporal confounding, potentially impacting the study’s validity. This heterogeneity among SCD patients, which includes variations in disease severity, concurrent treatments, and genetic differences, may have contributed to the variability in outcomes observed.
Further studies with larger, multicenter cohorts and detailed evaluations of clinical and genetic factors are warranted to validate our findings. Future research should also include patient-reported outcomes, such as pain scores and quality of life measures, as well as long-term safety assessments, to provide a more comprehensive evaluation of L-glutamine’s efficacy and safety in SCD patients.
5. Conclusions
L-glutamine treatment in our cohort of sickle cell disease patients did not result in statistically significant improvements in the frequency of VOCs, although some numerical trends were observed. Given the small sample size and limited statistical power, these findings should be interpreted as exploratory rather than definitive. Further well-powered, multicenter studies are warranted to validate the potential benefits of L-glutamine in this patient population.
Author Contributions
Conceptualization, S.T., A.A. (Atika Alharbi), M.K., A.A. (Aeshah AlAzmi), S.A. (Sultan Almutairi), and N.E.; methodology, S.T., A.A. (Atika Alharbi), M.K., A.A. (Aeshah AlAzmi), S.A. (Sultan Almutairi), and N.E.; project administration, S.T.; formal analysis, A.A. (Atika Alharbi), and S.A. (Sultan Alotaibi); validation, S.T. and A.A. (Atika Alharbi); writing—original draft, S.T.; writing—review and editing, S.T., A.A. (Atika Alharbi), M.K., A.A. (Aeshah AlAzmi), S.A. (Sultan Almutairi), and N.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was conducted in accordance with relevant ethical guidelines and approved by the Institutional Review Board of King Abdullah International Medical Research Center (KAIMRC) (protocol code NRJ22J/165/06, approved in 2022). Each patient was assigned a case report form (CRF) and a participant number, which were stored in a secure, separate location accessible only to the principal investigator (PI). Participants’ privacy and confidentiality were ensured, as no personal identifiers were included in the CRFs. All data, both hard and soft copies, were stored within MNGHA premises and were accessible only to the research team. Patient identities were protected by assigning identification codes, and medical record numbers were not used.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article.
Acknowledgments
The authors would like to acknowledge the funding (internal) and support provided by King Abdullah International Medical Research Center (KAIMRC) for this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Correction Statement
This article has been republished with a minor correction to the correspondence contact information. This change does not affect the scientific content of the article.
Abbreviations
The following abbreviations are used in this manuscript:
| CI | Confidence Interval |
| FDA | Food and Drug Administration |
| Hb | Hemoglobin |
| HbS | Hemoglobin S |
| HRQOL | Health-Related Quality of Life |
| IQR | Interquartile Range |
| LDH | Lactate Dehydrogenase |
| LOS | Length of Stay |
| NAD | Nicotinamide Adenine Dinucleotide |
| NUPCO | National Unified Procurement Company |
| PO | Oral |
| RBC | Red Blood Cell |
| SCD | Sickle Cell Disease |
| SFDA | Saudi Food and Drug Administration |
| SD | Standard Deviation |
| VOC | Vaso-Occlusive Crisis |
| P&T | Pharmacy and Therapeutic |
References
- Pecker, L.H.; Lanzkron, S. Sickle cell disease. Ann. Intern. Med. 2021, 174, ITC1–ITC16. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Bhor, M.; Xie, L.; Paulose, J.; Yuce, H. Sickle cell disease complications: Prevalence and resource utilization. PLoS ONE 2019, 14, e0214355. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Sickle Cell Disease (SCD): Data & Statistics; CDC: Atlanta, GA, USA, 2022. Available online: https://www.cdc.gov/sickle-cell/data/ (accessed on 18 April 2022).
- Ministry of Health (Saudi Arabia), Health Awareness: World Sickle Cell Day; Ministry of Health: Riyadh, Saudi Arabia. Available online: https://www.moh.gov.sa/en/HealthAwareness/healthDay/2019/Pages/HealthDay-2019-06-19.aspx (accessed on 18 April 2022).
- Ogu, U.O.; Badamosi, N.U.; Camacho, P.E.; Freire, A.X.; Adams-Graves, P. Management of sickle cell disease complications beyond acute chest syndrome. J. Blood Med. 2021, 25, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Brandow, A.M.; Carroll, C.P.; Creary, S.; Edwards-Elliott, R.; Glassberg, J.; Hurley, R.W.; Kutlar, A.; Seisa, M.; Stinson, J.; Strouse, J.J.; et al. American Society of Hematology 2020 guidelines for sickle cell disease: Management of acute and chronic pain. Blood Adv. 2020, 4, 2656–2701. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Ahmad, A.; Chaudry, H.; Aiman, W.; Aamir, S.; Anwar, M.Y.; Khan, A. Efficacy and safety of recently approved drugs for sickle cell disease: A review of clinical trials. Exp. Hematol. 2020, 92, 11–18.e1. [Google Scholar] [CrossRef]
- Salinas Cisneros, G.; Thein, S.L. Recent advances in the treatment of sickle cell disease. Front. Physiol. 2020, 11, 435. [Google Scholar] [CrossRef]
- Novartis. European Commission (EC) Adopts Decision Endorsing CHMP Recommendation to Revoke Conditional Marketing Authorization of Adakveo® (crizanlizumab); Novartis: Basel, Switzerland, 2023. Available online: https://www.novartis.com/news/european-commission-ec-adopts-decision-endorsing-chmp-recommendation-revoke-conditional-marketing-authorization-adakveo-crizanlizumab (accessed on 18 May 2025).
- Reuters. Pfizer Withdraws Sickle Cell Disease Treatment from All Markets. 2024. Available online: https://www.reuters.com/business/healthcare-pharmaceuticals/pfizer-withdraws-sickle-cell-disease-treatment-all-markets-2024-09-25/ (accessed on 19 May 2025).
- Sadaf, A.; Quinn, C.T. L-glutamine for sickle cell disease: Knight or pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef]
- Salman, E.K.; Haymond, M.W.; Bayne, E.; Sager, B.K.; Wiisanen, A.D.; Pitel, P.A.; Darmaun, D. Protein and energy metabolism in prepubertal children with sickle cell anemia. Pediatr. Res. 1996, 40, 34–40. [Google Scholar] [CrossRef]
- Nur, E.; Biemond, B.J.; Otten, H.M.; Brandjes, D.P.; Schnog, J.J.; CURAMA Study Group. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am. J. Hematol. 2011, 86, 484–489. [Google Scholar] [CrossRef]
- Niihara, Y.; Macan, H.; Eckman, J.R.; Koh, H.; Cooper, M.L.; Ziegler, T.R.; Razon, R.; Tanaka, K.R.; Stark, C.W.; Johnson, C.S. L-Glutamine therapy reduces hospitalization for sickle cell anemia and sickle β-thalassemia patients at six months–a phase II randomized trial. Clin. Pharmacol. Biopharm. 2014, 3, 1000116. [Google Scholar] [CrossRef]
- Niihara, Y.; Zerez, C.R.; Akiyama, D.S.; Tanaka, K.R. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am. J. Hematol. 1998, 58, 117–121. [Google Scholar] [CrossRef]
- Yassin, M.A. Evidence and Gaps in Clinical Outcomes of L-Glutamine in Sickle Cell Disease: A Systematic Literature Review Highlighting Insights from Clinical Trials and Real-World Studies. Blood 2024, 144, 5314. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A phase 3 trial of l-glutamine in sickle cell disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Drugs@FDA; FDA: Silver Spring, MD, USA, 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208587Orig1s000TOC.cfm (accessed on 20 April 2022).
- Maha Mohammed, A. Sickle cell disease in Saudi Arabia: A challenge or not. J. Epidemiol. Glob. Health 2017, 7, 99–101. [Google Scholar] [CrossRef]
- Jastaniah, W.; Al Zayed, A.H.; Al Saeed, H.H.; Al Darwish, M.; Albagshi, M.; Malhan, H.; Tarawah, A.; Al Manea, A.; Alhazmi, I.; Qari, M.; et al. Evidence Gaps in the Management of Patients with Sickle Cell Disease (SCD) by Non-specialist Healthcare Professionals (HCPs): Results from the Real-World Assessment Survey for SCD in Saudi (ROARS). Blood 2022, 140 (Suppl. S1), 5133–5134. [Google Scholar] [CrossRef]
- Abu Esba, L.C.; Almodaimegh, H.; Ahmed Khan, M.; Yousef, C.C.; Al-Abdulkarim, H.; Al Aklabi, A.A.; Al Harbi, M. A Formulary Management Group Consensus. Glob. J. Qual. Saf. Healthc. 2024, 7, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Alkhudair, N.; Howaidi, J.; Alnuhait, M.; Alshamrani, M.; Khan, M.; Alharbi, A.; Alnajjar, F.; Bajnaid, E.; Almodaheem, H.; Alhowimel, M.; et al. Revitalizing oncology medications access in Saudi Arabia: Current challenges and recommendations by the Saudi Oncology Pharmacy Assembly. J. Oncol. Pharm. Pract. 2025, 31, 245–250. [Google Scholar] [CrossRef]
- Emmaus Medical, Inc. Endari (L-glutamine oral powder): Full Prescribing Information. [Place Unknown]: Emmaus Medical, Inc. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208587s000lbl.pdf (accessed on 20 April 2022).
- Elenga, N.; Loko, G.; Etienne-Julan, M.; Al-Okka, R.; Adel, A.M.; Yassin, M.A. Real-World data on efficacy of L-glutamine in preventing sickle cell disease-related complications in pediatric and adult patients. Front. Med. 2022, 9, 931925. [Google Scholar] [CrossRef]
- Dhaliwal, G.; Cornett, P.A.; Tierney, L.M., Jr. Hemolytic anemia. Am. Fam. Physician 2004, 69, 2599–2607. [Google Scholar]
- Carden, M.A.; Fasano, R.M.; Meier, E.R. Not all red cells sickle the same: Contributions of the reticulocyte to disease pathology in sickle cell anemia. Blood Rev. 2020, 40, 100637. [Google Scholar] [CrossRef]
- De Haan, L.D.; Werre, J.M.; Ruben, A.T.; Huls, A.H.; De Gier, J.; Staal, G.E. Reticulocyte crisis after splenectomy: Evidence for delayed red cell maturation? Eur. J. Haematol. 1988, 41, 74–79. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}