Genetic Diversity among Selected Medicago sativa Cultivars Using Inter-Retrotransposon-Amplified Polymorphism, Chloroplast DNA Barcodes and Morpho-Agronomic Trait Analyses


Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. Extraction, Purification, and Quantification of Genomic DNA of M. sativa Cultivars
2.3. IRAP Primer and IRAP Marker PCR Amplification
2.4. DNA Barcoding of mat K and trn H Plastid Genes
2.5. Data Analysis
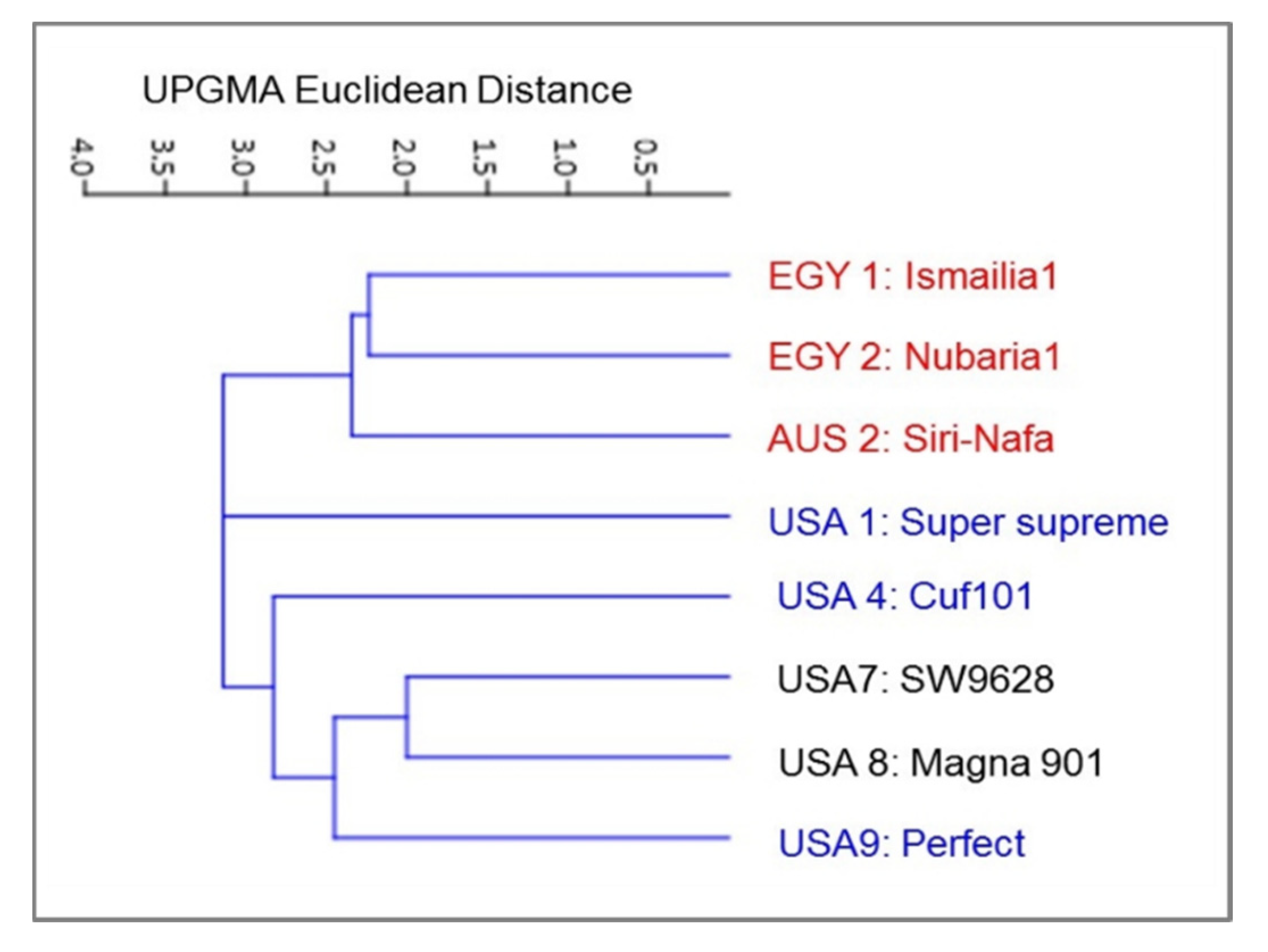
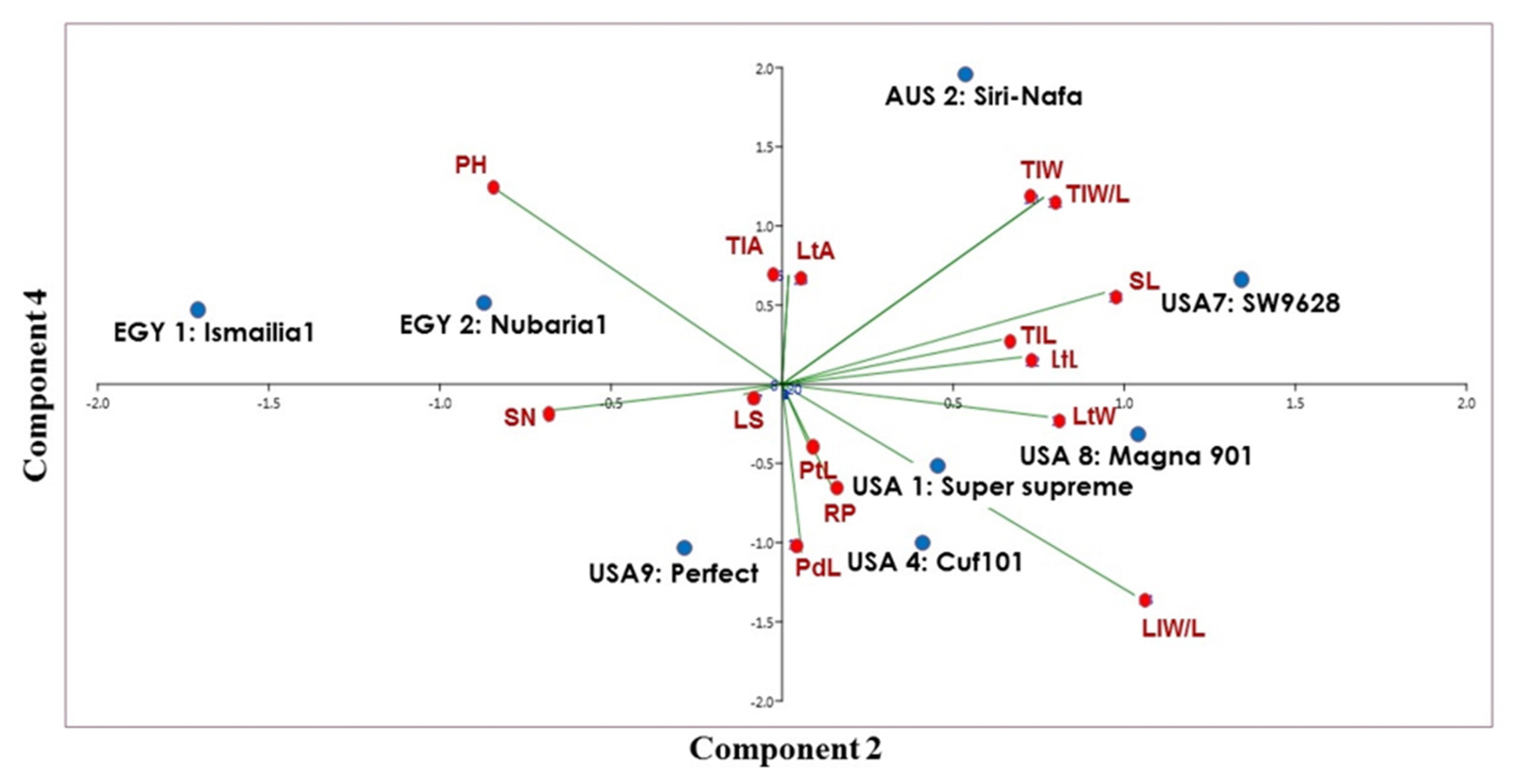
3. Results
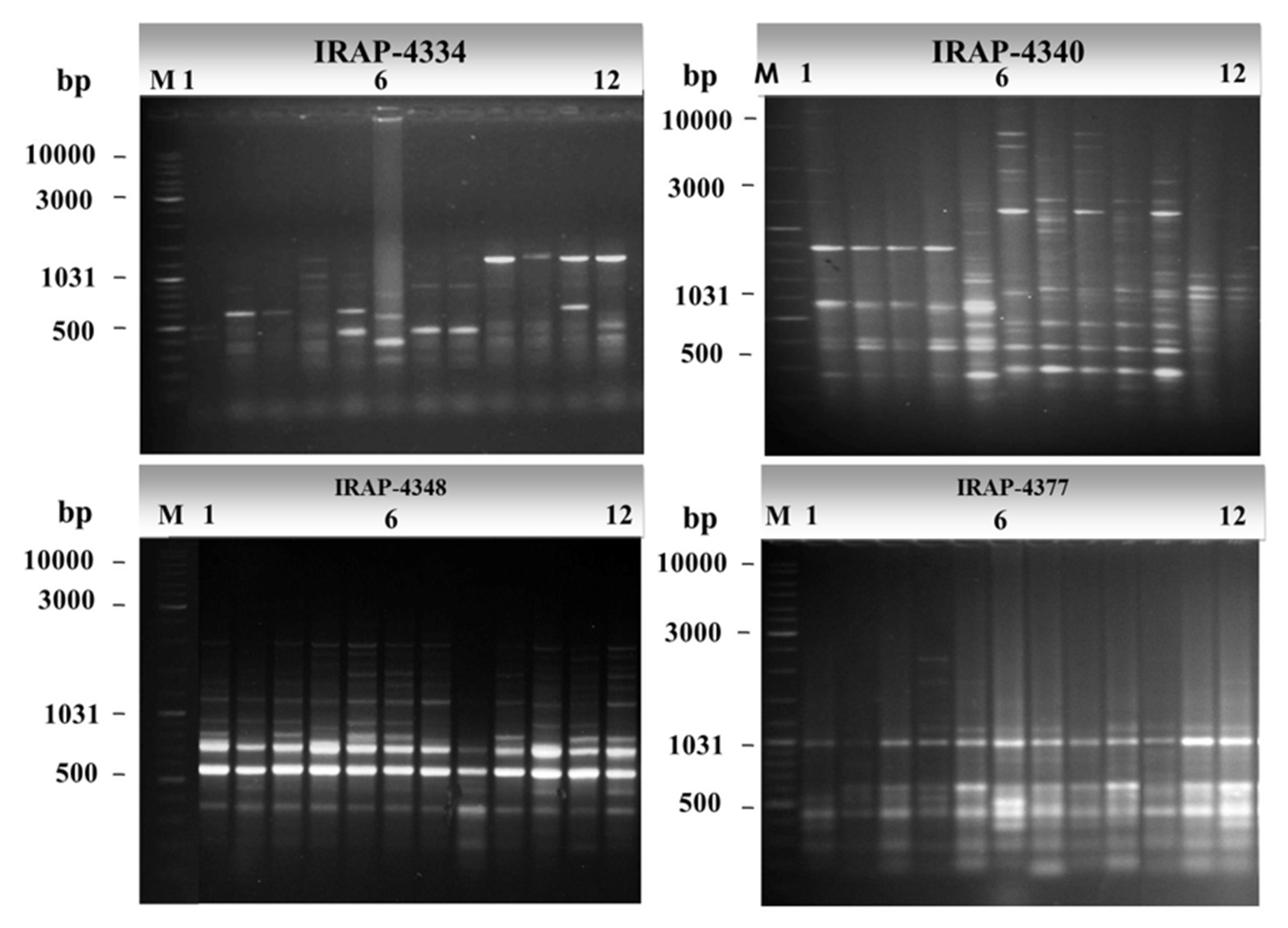
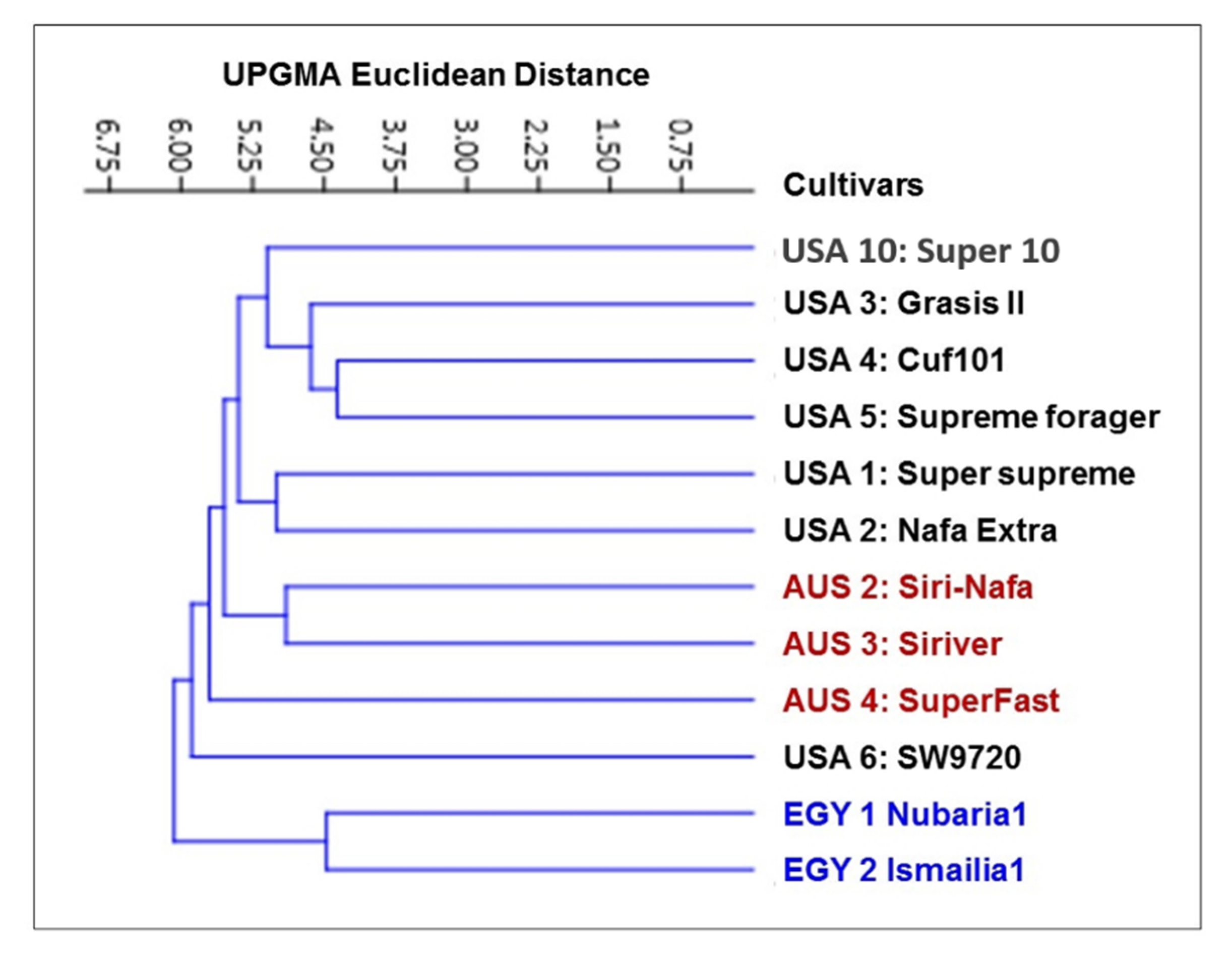
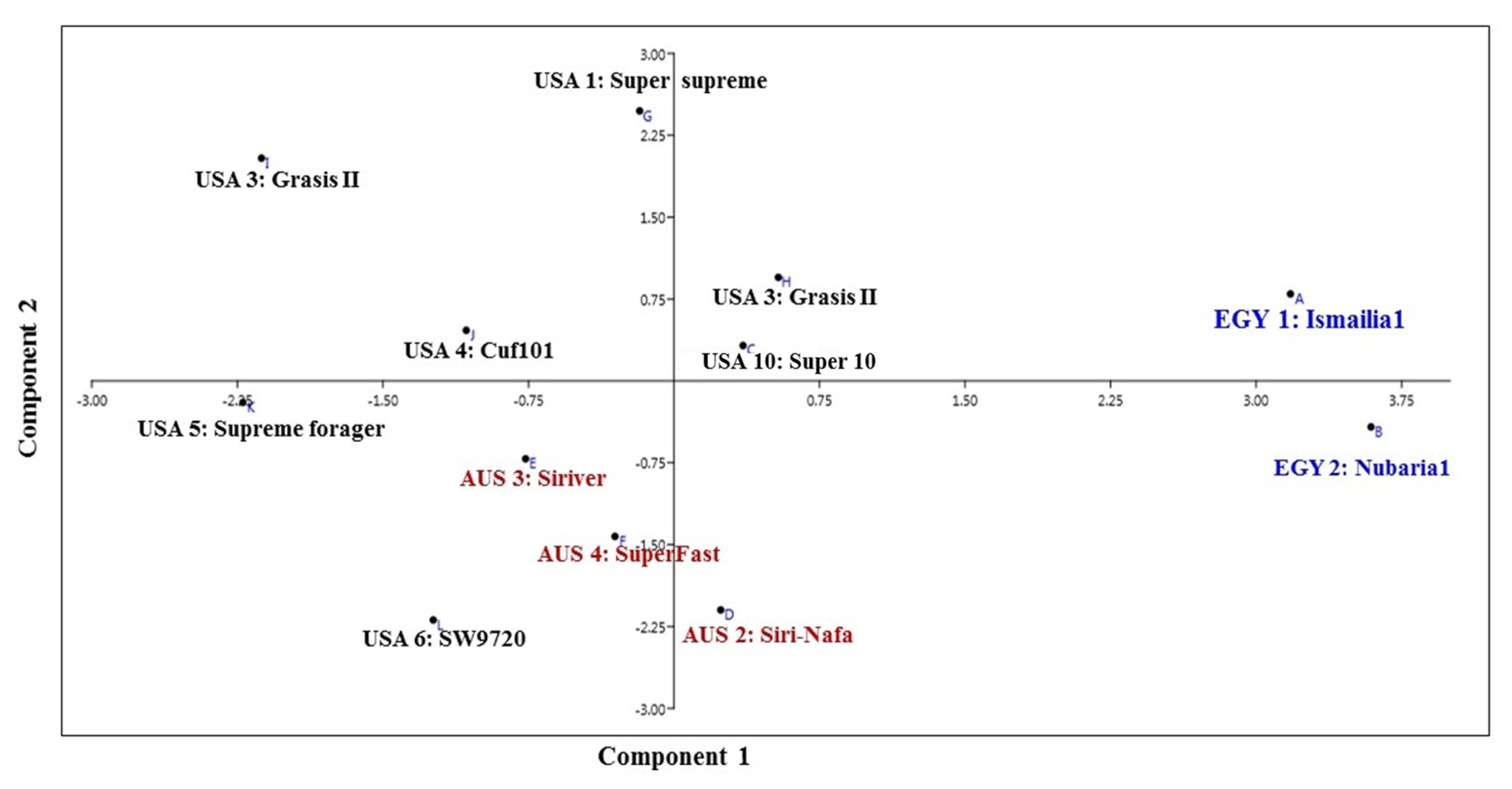
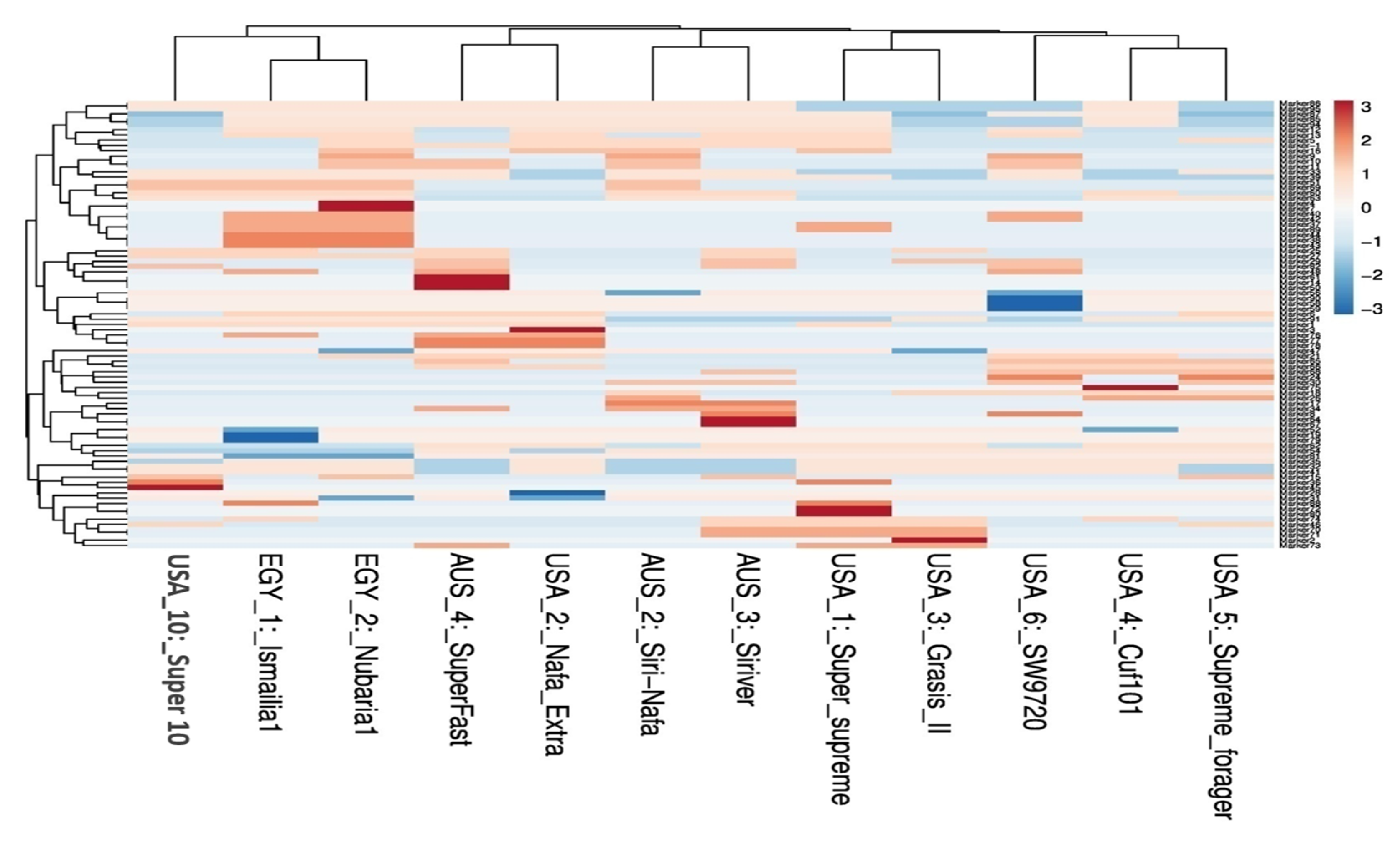
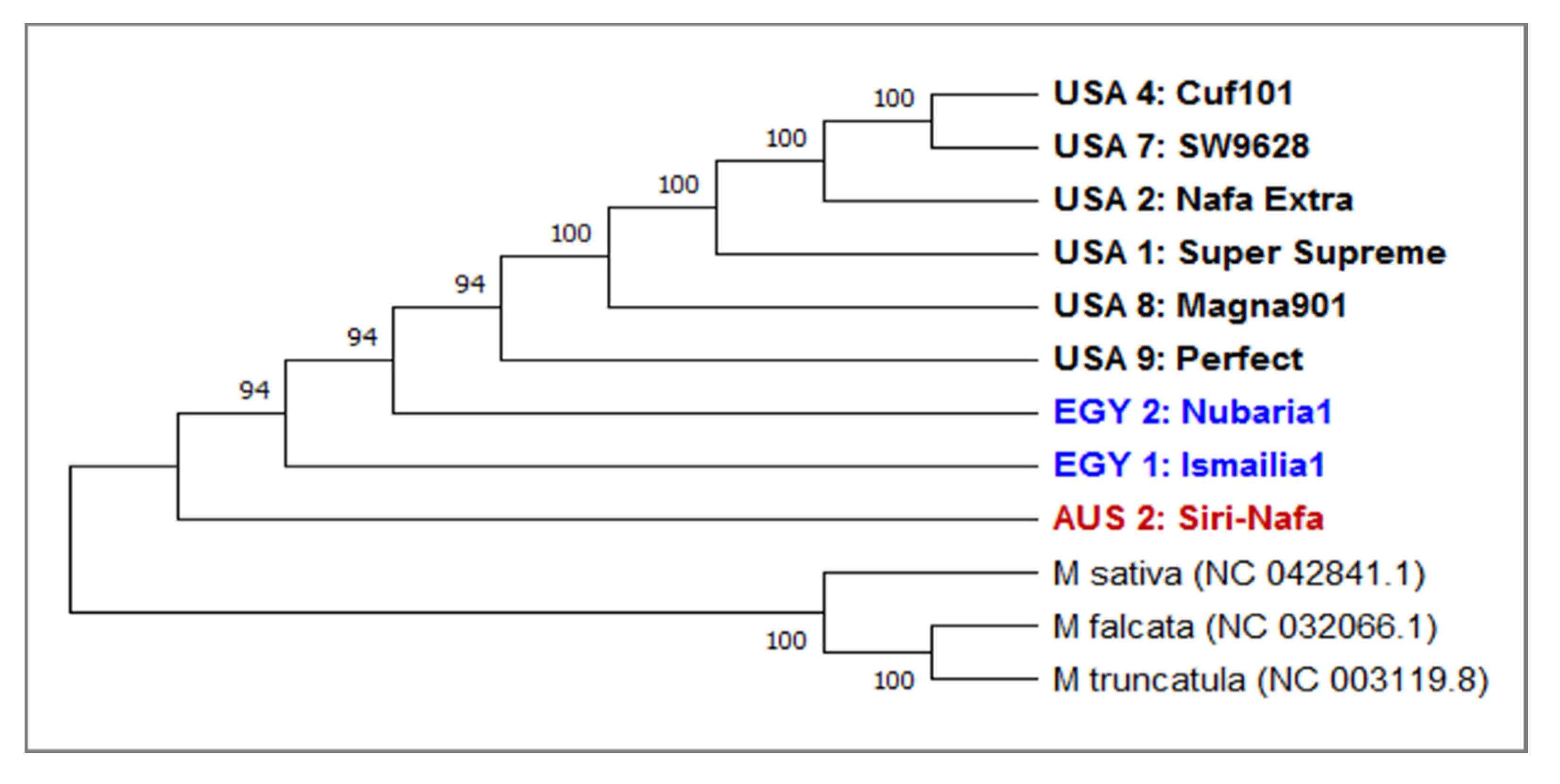
3.1. Molecular Characterization and Genetic Relationships as Revealed by IRAP Markers
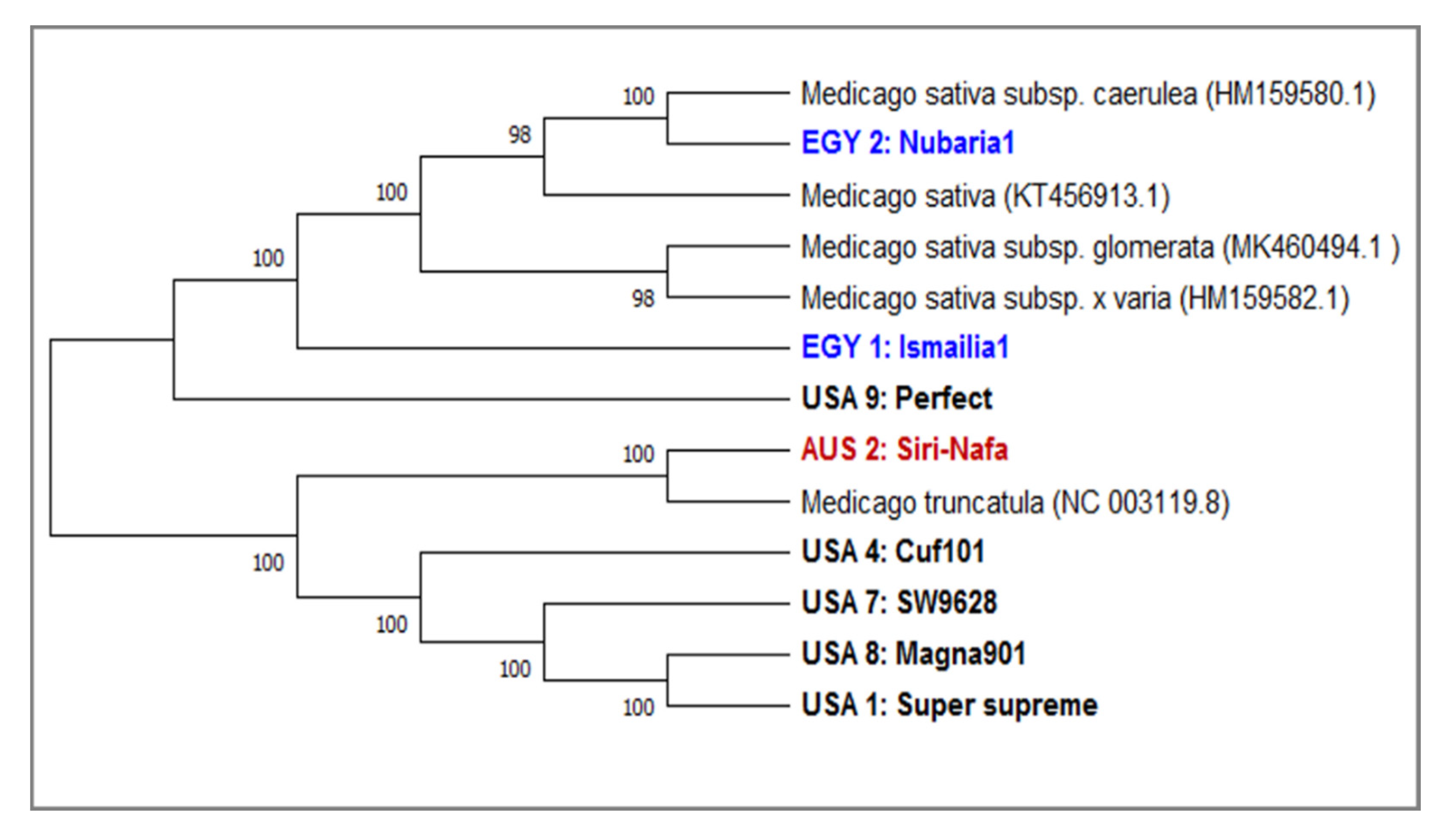
3.2. DNA Barcoding Loci of matK and trnH Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suttie, J.M. Alfalfa and Relatives: Evolution and Classification of Medicago. By E. Small. Wallingford, UK: CABI (2011), pp. 727, £110.00. Exp. Agric. 2012, 48, 308–309. [Google Scholar] [CrossRef]
- Li, X.; Brummer, E.C. Applied Genetics and Genomics in Alfalfa Breeding. Agronomy 2012, 2, 40–61. [Google Scholar] [CrossRef]
- Blondon, D.; Marie, S.; Brown, A. Kondorosi, Genome size and base composition in Medicago sativa and M. truncatula species. Genome 1994, 37, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, F.; Brummer, E.C. Handbook of Plant Breeding: Fodder Crops and Amenity Grasses; Springer: New York, NY, USA, 2010. [Google Scholar]
- Annicchiarico, P. Diversity, genetic structure, distinctness and agronomic value of Italian lucerne (Medicago sativa L.) landraces. Euphytica 2006, 148, 269–282. [Google Scholar] [CrossRef]
- Tucak, M.; Popović, S.; Čupic, T.; Šimic, G.; Gantner, R.; Meglič, V. Evaluation of alfalfa germplasm collection by multivariate analysis based on phenotypic traits. Rom. Agric. Res. 2009, 26, 47–51. [Google Scholar]
- Tucak, M.; Popović, S.; Čupić, T.; Grljušić, S.; Meglič, V.; Jurković, Z. Efficiency of phenotypic and DNA markers for a genetic diversity study of alfalfa. Гeнeтикa 2010, 46, 1314–1319. [Google Scholar] [CrossRef]
- Bornet, M. Branchard, Nonanchored Inter Simple Sequence Repeat (ISSR) markers: Reproducible and specific tools for genome fingerprinting. Plant Mol. Biol. Rep. 2001, 19, 209–215. [Google Scholar] [CrossRef]
- Semagn, K.; Bjørnstad, Å.; Ndjiondjop, M.N. An overview of molecular marker methods for plants. Afr. J. Biotechnol. 2006. [Google Scholar] [CrossRef]
- Badr, A. Molecular approaches to plant systematic and evolution. Taeckholmia 2008, 28, 127–167. [Google Scholar]
- El-Shazly, H.H.; HAhmed, I.S.; Hamouda, M.M.; Badr, A. Genetic diversity and population structure of the medicinal plant Achillea fragrantissima (Forssk.) Sch. Bip. in the mountains of South Sinai, Egypt. Plant Gene. 2020, 21, 100212. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Start Codon Targeted (SCoT) Polymorphism: A Simple, Novel DNA Marker Technique for Generating Gene-Targeted Markers in Plants. Plant Mol. Biol. Rep. 2009, 27, 86–93. [Google Scholar] [CrossRef]
- Mardi, M.; Naghavi, M.R.; Pirseyedi, S.M.; Alamooti, M.K.; Monfared, S.R.; Ahkami, A.H.; Omidbakhsh, M.A.; Alavi, N.S.; Shanjani, P.S.; Katsiotis, A. Comparative assessment of SSAP, AFLP and SSR markers for evaluation of genetic diversity of durum wheat (Triticum turgidum L. var. durum). J. Agric. Sci. Technol. 2011, 13, 905–920. [Google Scholar]
- Bagavathiannan, M.V.; Julier, B.; Barre, P.; Gulden, R.H.; van Acker, R.C. Genetic diversity of feral alfalfa (Medicago sativa L.) populations occurring in Manitoba, Canada and comparison with alfalfa cultivars: An analysis using SSR markers and phenotypic traits. Euphytica 2010, 173, 419–432. [Google Scholar] [CrossRef]
- Robarts, D.W.H.; Wolfe, A.D. Sequence-Related Amplified Polymorphism (SRAP) Markers: A Potential Resource for Studies in Plant Molecular Biology. Appl. Plant Sci. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Al-Faifi, S.A.; Migdadi, H.; Al-Doss, A.; Ammar, M.H.; El-Harty, E.H.; Khan, M.A.; Muhammad, J.M.; Alghamdi, S.S. Morphological and molecular genetic variability analyses of Saudi lucerne (Medicago sativa L.) landraces. Crop. Pasture Sci. 2013, 64, 137–146. [Google Scholar] [CrossRef]
- Ben Rhouma, H.; Taski-Ajdukovic, K.; Zitouna, N.; Sdouga, D.; Milic, D.; Trifi-Farah, N. Assessment of the genetic variation in alfalfa genotypes using SRAP markers for breeding purposes. Chil. J. Agric. Res. 2017, 77, 332–339. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kalendar, R.; Schulman, A.H. Transposon-Based Tagging: IRAP, REMAP, and iPBS. Adv. Struct. Saf. Stud. 2013, 1115, 233–255. [Google Scholar] [CrossRef]
- Mandoulakani, A.B.; Sadigh, P.; Azizi, H.; Piri, Y.; Nasri, S. Comparative assessment of IRAP, REMAP, ISSR, and SSR markers for evaluation of genetic diversity of alfalfa (Medicago sativa L.). J. Agric. Sci. Technol. 2015, 17, 999–1010. [Google Scholar]
- Baumel, A.; Ainouche, M.L.; Kalendar, R.; Schulman, A.H. Retrotransposons and genomic stability in populations of the young allopolyploid species Spartina anglica C.E. Hubbard (Poaceae). Mol. Biol. Evol. 2002, 19, 1218–1227. [Google Scholar] [CrossRef]
- Nasri, S.; Mandoulakani, B.A.; Darvishzadeh, R.; Bernousi, I. Retrotransposon Insertional Polymorphism in Iranian Bread Wheat Cultivars and Breeding Lines Revealed by IRAP and REMAP Markers. Biochem. Genet. 2013, 51, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Farouji, A.E.; Khodayari, H.; Saeidi, H.; Rahiminejad, M.R. Genetic diversity of diploid Triticum species in Iran assessed using inter-retroelement amplified polymorphisms (IRAP) markers. Biology 2015, 70, 52–60. [Google Scholar] [CrossRef]
- Taheri, M.T.; Alavi-Kia, S.S.; Mohammadi, S.A.; Vahed, M.M. Assessment of genetic diversity and relationships among Triticum urartu and Triticum boeoticum populations from Iran using IRAP and REMAP markers. Genet. Resour. Crop. Evol. 2018, 65, 1867–1878. [Google Scholar] [CrossRef]
- Guo, Y.; Zhai, L.; Long, H.; Chen, N.; Gao, C.; Ding, Z.; Jin, B. Genetic diversity of Bletilla striata assessed by SCoT and IRAP markers. Hereditas 2018, 155, 35. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; Dewaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and Using a Plant DNA Barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Singer, G.A.; Clare, E.L.; Hebert, P.D.N. Design and applicability of DNA arrays and DNA barcodes in biodiversity monitoring. BMC Biol. 2007, 5, 24. [Google Scholar] [CrossRef]
- Pei, N.; Chen, B.; Kress, W.J. Advances of Community-Level Plant DNA Barcoding in China. Front. Plant Sci. 2017, 8, 225. [Google Scholar] [CrossRef]
- Whitlock, B.A.; Hale, A.M.; Groff, P.A. Intraspecific Inversions Pose a Challenge for the trnH-psbA Plant DNA Barcode. PLoS ONE 2010, 5, e11533. [Google Scholar] [CrossRef]
- Madesis, P.; Ganopoulos, I.; Ralli, P.; Tsaftaris, A. Barcoding the major Mediterranean leguminous crops by combining universal chloroplast and nuclear DNA sequence targets. Genet. Mol. Res. 2012, 11, 2548–2558. [Google Scholar] [CrossRef]
- Bondok, A. Using DNA Barcoding for Fingerprinting of Two Important Forage Crops Varieties (Alfalfa and Egyptian Clover). J. Agric. Chem. Biotechnol. 2019, 10, 195–201. [Google Scholar] [CrossRef]
- Riday, H.; Brummer, E.C. Morphological variation of Medicago sativa subsp. falcata genotypes and their hybrid progeny. Euphytica 2004, 138, 1–12. [Google Scholar] [CrossRef]
- Warburton, M.L.; Smith, S.E. Regional Diversity in Nondormant Alfalfas from India and the Middle East. Crop. Sci. 1993, 33, 852–858. [Google Scholar] [CrossRef]
- Rumbaugh, M.D.; Graves, W.L.; Caddel, J.L.; Mohammad, R.M. Variability in a Collection of Alfalfa Germplasm from Morocco. Crop. Sci. 1988, 28, 605–609. [Google Scholar] [CrossRef]
- Rumbaugh, M.D. Plant Introductions: The Foundation of North American Forage Legume Cultivar Development; John Wiley & Sons, Inc.: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Riday, H.; Brummer, E.C.; Moore, K.J. Heterosis of Forage Quality in Alfalfa. Crop. Sci. 2002, 42, 1088–1093. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harb: New York, NY, USA, 1988. [Google Scholar]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Moawed, M.M. Evaluation of morphological and anatomical characters for discrimination and verification of some Medicago sativa (L.) Cultivars. Indian J. Agric. Res. 2016, 50. [Google Scholar] [CrossRef]
- Ghislain, M.; Zhang, D.; Fajardo, D.; Huamán, Z.; Hijmans, R. Marker-assisted sampling of the cultivated Andean potato Solanum phureja collection using RAPD markers. Genet. Resour. Crop. Evol. 1999, 46, 547–555. [Google Scholar] [CrossRef]
- Gilbert, J.E.; Lewis, R.V.; Wilkinson, M.J.; Caligari, P.D.S. Developing an appropriate strategy to assess genetic variability in plant germplasm collections. Theor. Appl. Genet. 1999, 98, 1125–1131. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron 2001, 4, 1–9. [Google Scholar]
- Turhan Serttaş, P.; Özcan, T. Intraspecific variations studied by ISSR and IRAP markers in mastic tree (Pistacia lentiscus L.) from Turkey, Trak. Univ. J. Nat. Sci. 2018. [Google Scholar] [CrossRef][Green Version]
- Kashkush, K.; Feldman, M.; Levy, A. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 2003, 33, 102–106. [Google Scholar] [CrossRef]
- Mansour, A. Utilization of genomic retrotransposons as cladistic markers. J. Cell Mol. Biol. 2008, 7, 17–28. [Google Scholar]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.N.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 2010, 106, 520–530. [Google Scholar] [CrossRef]
- Mengoni, A.; Gori, A.; Bazzicalupo, M. Use of RAPD and microsatellite (SSR) variation to assess genetic relationships among populations of tetraploid alfalfa, Medicago sativa. Plant Breed. 2000, 119, 311–317. [Google Scholar] [CrossRef]
- Falahati-Anbaran, M.; Habashi, A.A.; Esfahany, M.; Mohammadi, S.A.; Ghareyazie, B. Population genetic structure based on SSR markers in alfalfa (Medicago sativa L.) from various regions contiguous to the centres of origin of the species. J. Genet. 2007, 86, 59–63. [Google Scholar] [CrossRef]
- Inglis, P.W.; Mata, L.R.; Da Silva, M.J.; Vieira, R.F.; Alves, R.D.B.; Silva, D.B.; Azevedo, V.C. DNA Barcoding for the Identification of Phyllanthus Taxa Used Medicinally in Brazil. Planta Med. 2018, 84, 1300–1310. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, Y.; Wang, X.; Wei, X.; Han, J. DNA Mini-Barcoding: A Derived Barcoding Method for Herbal Molecular Identification. Front. Plant Sci. 2019, 10, 987. [Google Scholar] [CrossRef]
- Loumerem, M.; Ferchichi, A.; Haddad, M.; Rahim, M.A.A.; Hajjaji, H. Collection and evaluation of lucerne (Medicago sativa L.) germplasm from oases of Tunisia. Genet. Resour. Crop. Evol. 2007, 54, 1645–1651. [Google Scholar] [CrossRef]
- N.A.F.A. (NAFA) Winter Survival, Fall Dormancy & Pest Resistance Ratings for Alfalfa Varieties, 2014th ed. 2014. Available online: www.alfalfa.org (accessed on 1 May 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Origin | Code # | Cultivar Name | matK Genebank Accession | trnH Genebank Accession |
|---|---|---|---|---|---|
| 1 | Egypt (EGY) | EGY1 | Ismailia1 * ARC | MN509171 | MN509787 |
| 2 | EGY1 | Nubaria1 * ARC | MN509788 | MN610248 | |
| 3 | Australia (AUS) | AUS2 | Siri-Nafa * Australia | MN509173 | MN610246 |
| 4 | AUS3 | Siriver Australia | NA | NA | |
| 5 | AUS4 | SuperFast Saudi Arabia | NA | NA | |
| 6 | United States of America (USA) | USA1 | Super-supreme * California | MN509175 | MN509790 |
| 7 | USA2 | Nafa Extra USA | NA | NA | |
| 8 | USA3 | Grasis II California | NA | NA | |
| 9 | USA4 | Cuf101 * California | MN509177 | MN610247 | |
| 10 | USA5 | Supreme forager California | NA | NA | |
| 11 | USA6 | SW9720 California | NA | NA | |
| 12 | USA7 × | SW9628 *- California | MN610249 | MN509792 | |
| 13 | USA8 × | Magna901 * Diary Land | MN509174 | MN509789 | |
| 14 | USA9 × | Perfect * California | MN509172 | MN610245 | |
| 15 | USA10 | Super10 California | NA | NA |
| No. | Primer Name | Sequence (5′–3′) | TNAs | MAs | PAs | % P | PIC | RP | EMR | MI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IRAP4352 | ACCCGGAAGGGCGGTTCATGCAA | 9 | 1 | 8 | 89 | 0.78 | 6.5 | 7.11 | 5.57 |
| 2 | IRAP 4334 | CCATGGCGAGCAGATGTGCT | 11 | 0 | 11 | 100 | 0.88 | 7 | 11 | 9.67 |
| 3 | IRAP 4348 | TTAGATGAAACCAACGATCCCAAGGCT | 13 | 3 | 10 | 77 | 0.52 | 16 | 7.69 | 4.02 |
| 4 | IRAP 4370 | ATGCCGTATTCTCAGCATCC | 14 | 0 | 14 | 100 | 0.81 | 10.2 | 14 | 11.41 |
| 5 | IRAP 4377 | CGTACCCTTTTAAGGGATCAAAACC | 9 | 2 | 7 | 78 | 0.53 | 10.8 | 5.44 | 2.91 |
| 6 | IRAP 4351 | CAGGCAAGAATGAGCGTCTC | 3 | 2 | 1 | 33 | 0.1 | 5.7 | 0.33 | 0.03 |
| 7 | IRAP 4340 | ATGGTTGTCGAAACTCCAGC | 21 | 0 | 21 | 100 | 0.83 | 14.2 | 21 | 17.36 |
| 8 | IRAP 4342 | GATTGCAAAGCCTATTTCGCTG | 4 | 0 | 4 | 100 | 0.59 | 4.3 | 4 | 2.34 |
| 9 | IRAP 4375 | ATCGCTCCGGGTGCCTAACAC | 4 | 2 | 2 | 50 | 0.28 | 6.7 | 1 | 0.28 |
| 10 | IRAP 3471 | ATCGCTCCGGGTGCCTAACAC | 5 | 0 | 5 | 100 | 0.61 | 5.5 | 5 | 3.06 |
| 11 | IRAP 4357 | TGACATTTGTGGCACTTTCTGGCGT | 4 | 2 | 2 | 50 | 0.28 | 6.7 | 1 | 0.28 |
| Total | 97 | 12 | 85 | NA | NA | NA | NA | NA | ||
| Mean | 7.72 | 79.73 | 0.56 | 8.50 | 7.05 | 5.17 | ||||
| Primer Code | Sequence | Product Size | Reference |
|---|---|---|---|
| trnH-F | 5′-GTTATGCATGAACGTAATGCTC-3′ | 700 bp | [31] |
| trnH-R | 5′-CGCGCATGGTGGATTCACAATCC-3′ | [32] | |
| matK-472F | 5′-CCCRTYCATCTGGAAATCTTGGTTC-3′ | 700 bp | [33] |
| matK-1248R | 5′-GCTRTRATAATGAGAAAGATTTCTGC-3′ |
| Gene Name | Aligned Length | Undetermined Characters | Missing Percent | Variable Sites | Proportion Variable Sites | Parsimony Informative Sites | Proportion Parsimony Informative | AT% | GC% |
|---|---|---|---|---|---|---|---|---|---|
| matk | 911 | 863 | 13.53 | 327 | 0.359 | 194 | 0.213 | 0.66 | 0.34 |
| trnH. | 755 | 521 | 8.63 | 31 | 0.041 | 4 | 0.005 | 0.64 | 0.36 |
| Characters and Their Abbreviations | EGY1: Ismailia1 | EGY2: Nubaria1 | AUS2: Siri-Nafa | USA1: Super Supreme | USA4: Cuf101 | USA7: SW9628 | USA 8: Magna 901 | USA9: Perfect |
|---|---|---|---|---|---|---|---|---|
| Resistance to pests: RP | sensitive | sensitive | resistant | sensitive | sensitive | sensitive | sensitive | sensitive |
| Plant height (cm): PH | ≥70 | ≥70 | <70 | ≥70 | ≥70 | <70 | <70 | <70 |
| Stem number: SN | <4 | <4 | <4 | ≥4 | ≥4 | <4 | <4 | <4 |
| Petiole length (mm): PtL | <20 | <20 | <20 | ≥20 | <20 | <20 | <20 | <20 |
| Petiole hairiness: PtH | glabrous | hairy | hairy | glabrous | glabrous | glabrous | hairy | glabrous |
| Terminal leaflet length (mm): TlL | <20 | ≥4 | <20 | ≥4 | <20 | ≥4 | <20 | <20 |
| Terminal leaflet width (mm): TlW | <8 | ≥8 | <8 | ≥8 | <8 | ≥8 | <8 | <8 |
| Terminal leaflet width/length: TlW/L | <0.4 | ≥0.4 | <0.4 | ≥0.4 | <0.4 | ≥0.4 | <0.4 | <0.4 |
| Lateral leaflet length (mm) LtL | ≥18 | ≥18 | ≥18 | <18 | <18 | ≥18 | ≥18 | ≥18 |
| Lateral leaflet width (mm): LtW | ≥5 | ≥5 | ≥5 | ≥5 | <5 | ≥5 | ≥5 | ≥5 |
| Lateral leaflet width/length: LlW/L | <0.32 | <0.32 | ≥0.32 | ≥0.32 | <0.32 | ≥0.32 | ≥0.32 | <0.32 |
| Terminal leaflet apex shape: TlA | mucronate | mucronate | mucronate | mucronate | mucronate | mucronate | mucronate | hooked |
| Leaf stipule shape: LS | hairy | leafy | leafy | leafy | leafy | leafy | hairy | leafy |
| Stipule length (mm): SL | ≥9 | <9 | <9 | <9 | <9 | <9 | ≥9 | <9 |
| Peduncle length (mm): PdL | <18 | ≥18 | <18 | <18 | <18 | ≥18 | ≥18 | ≥18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badr, A.; El-Sherif, N.; Aly, S.; Ibrahim, S.D.; Ibrahim, M. Genetic Diversity among Selected Medicago sativa Cultivars Using Inter-Retrotransposon-Amplified Polymorphism, Chloroplast DNA Barcodes and Morpho-Agronomic Trait Analyses. Plants 2020, 9, 995. https://doi.org/10.3390/plants9080995
Badr A, El-Sherif N, Aly S, Ibrahim SD, Ibrahim M. Genetic Diversity among Selected Medicago sativa Cultivars Using Inter-Retrotransposon-Amplified Polymorphism, Chloroplast DNA Barcodes and Morpho-Agronomic Trait Analyses. Plants. 2020; 9(8):995. https://doi.org/10.3390/plants9080995
Chicago/Turabian StyleBadr, Abdelfattah, Nahla El-Sherif, Sara Aly, Shafik D. Ibrahim, and Mohamed Ibrahim. 2020. "Genetic Diversity among Selected Medicago sativa Cultivars Using Inter-Retrotransposon-Amplified Polymorphism, Chloroplast DNA Barcodes and Morpho-Agronomic Trait Analyses" Plants 9, no. 8: 995. https://doi.org/10.3390/plants9080995
APA StyleBadr, A., El-Sherif, N., Aly, S., Ibrahim, S. D., & Ibrahim, M. (2020). Genetic Diversity among Selected Medicago sativa Cultivars Using Inter-Retrotransposon-Amplified Polymorphism, Chloroplast DNA Barcodes and Morpho-Agronomic Trait Analyses. Plants, 9(8), 995. https://doi.org/10.3390/plants9080995