Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics



Abstract

1. Introduction
2. Results
2.1. Identification of Organisms in the Mummified Peach Fruits by DNA Shotgun Sequencing and RNA-Sequencing
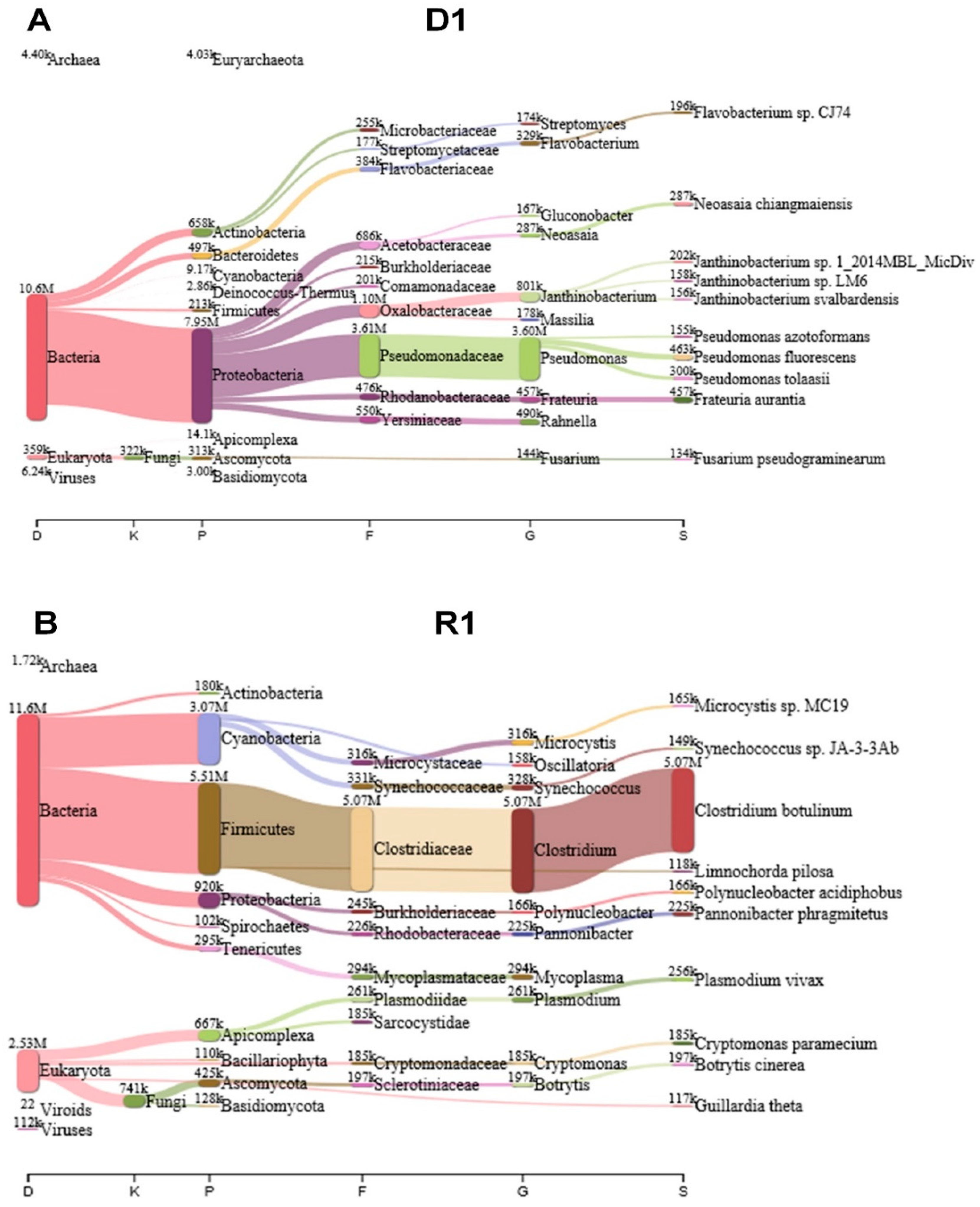
2.2. Taxonomic Classification Using KRAKEN 2 Program
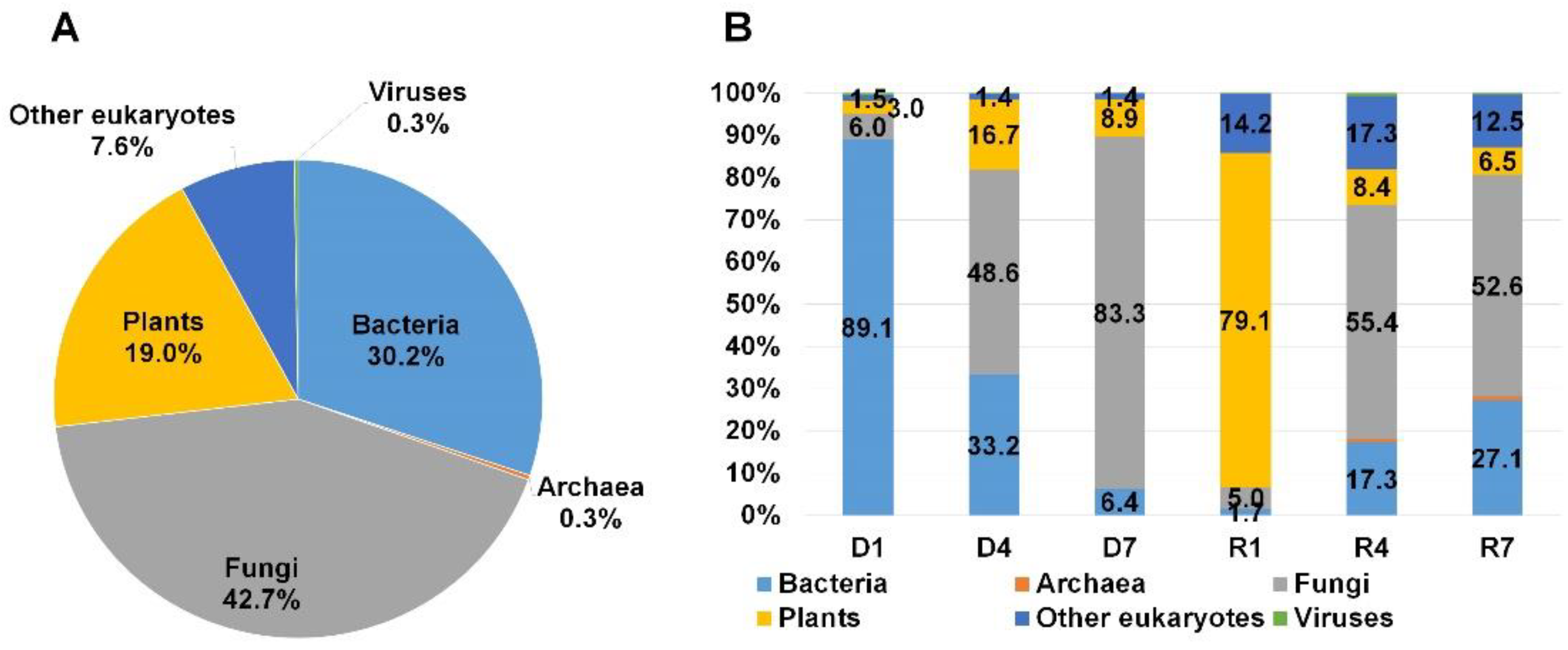
2.3. Classification of Identified Organisms on the Three Mummified Peach Fruits According to Domain
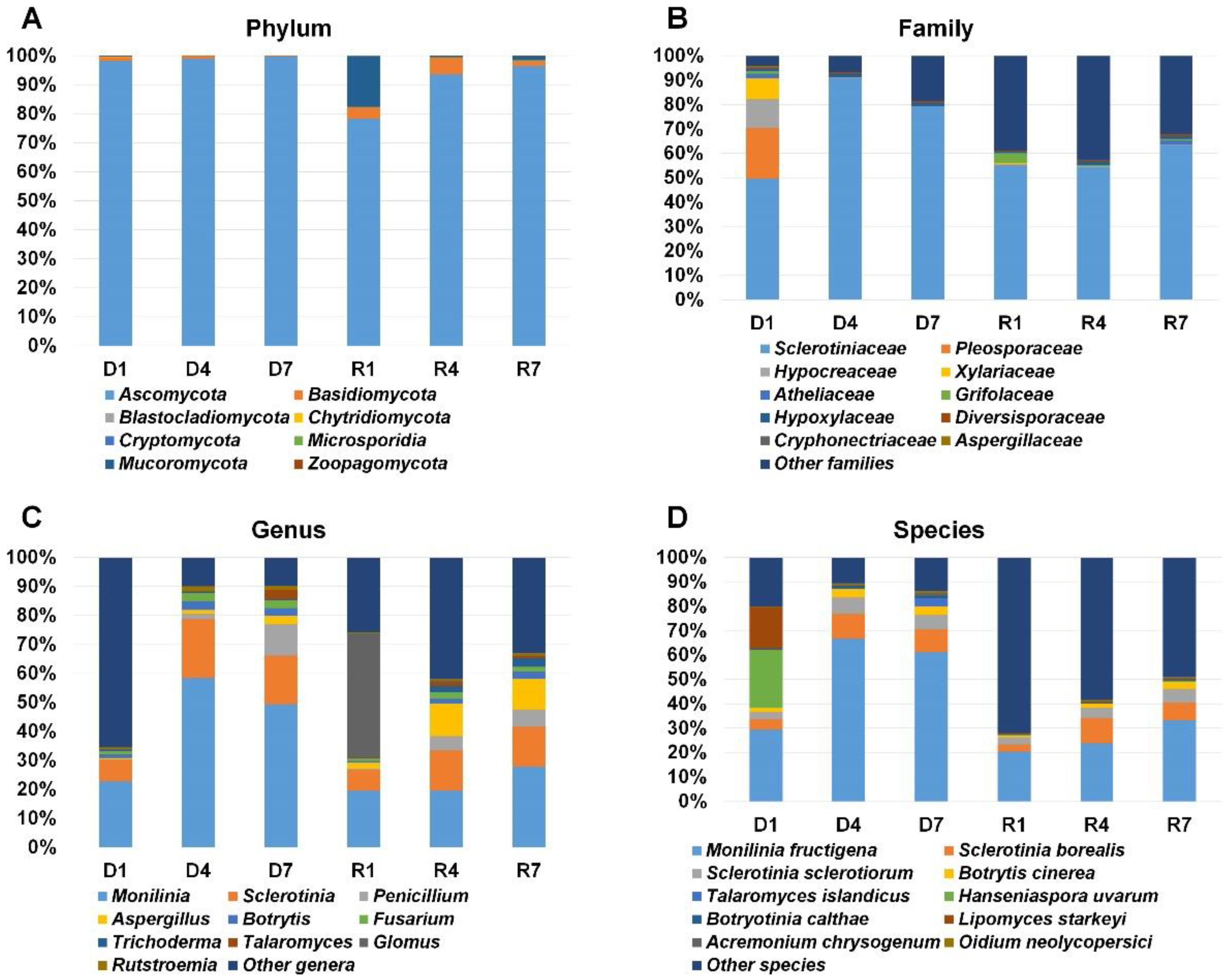
2.4. Classification of Identified Fungi According to Fungal Taxonomy
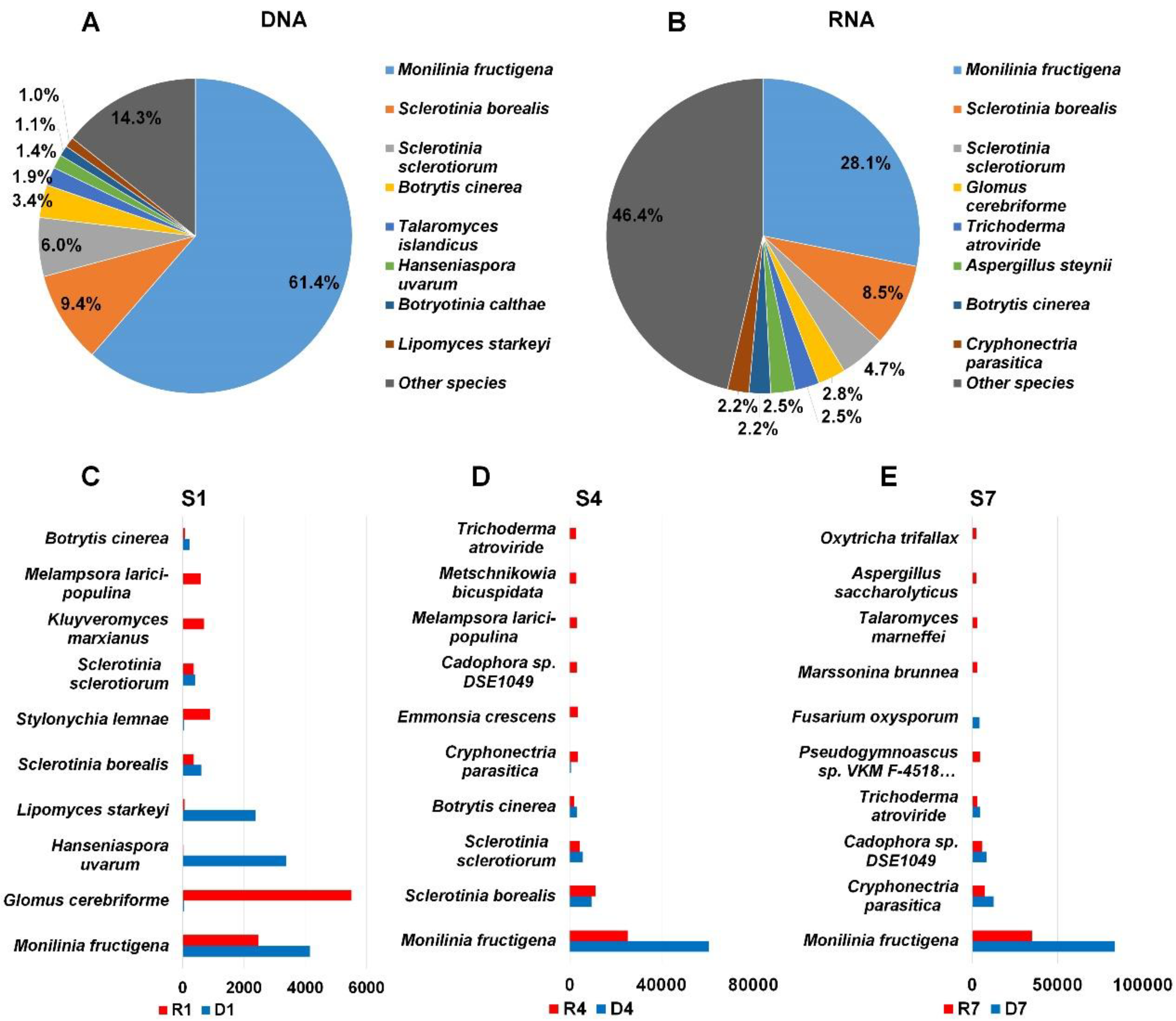
2.5. Major Fungal Species in Each Library
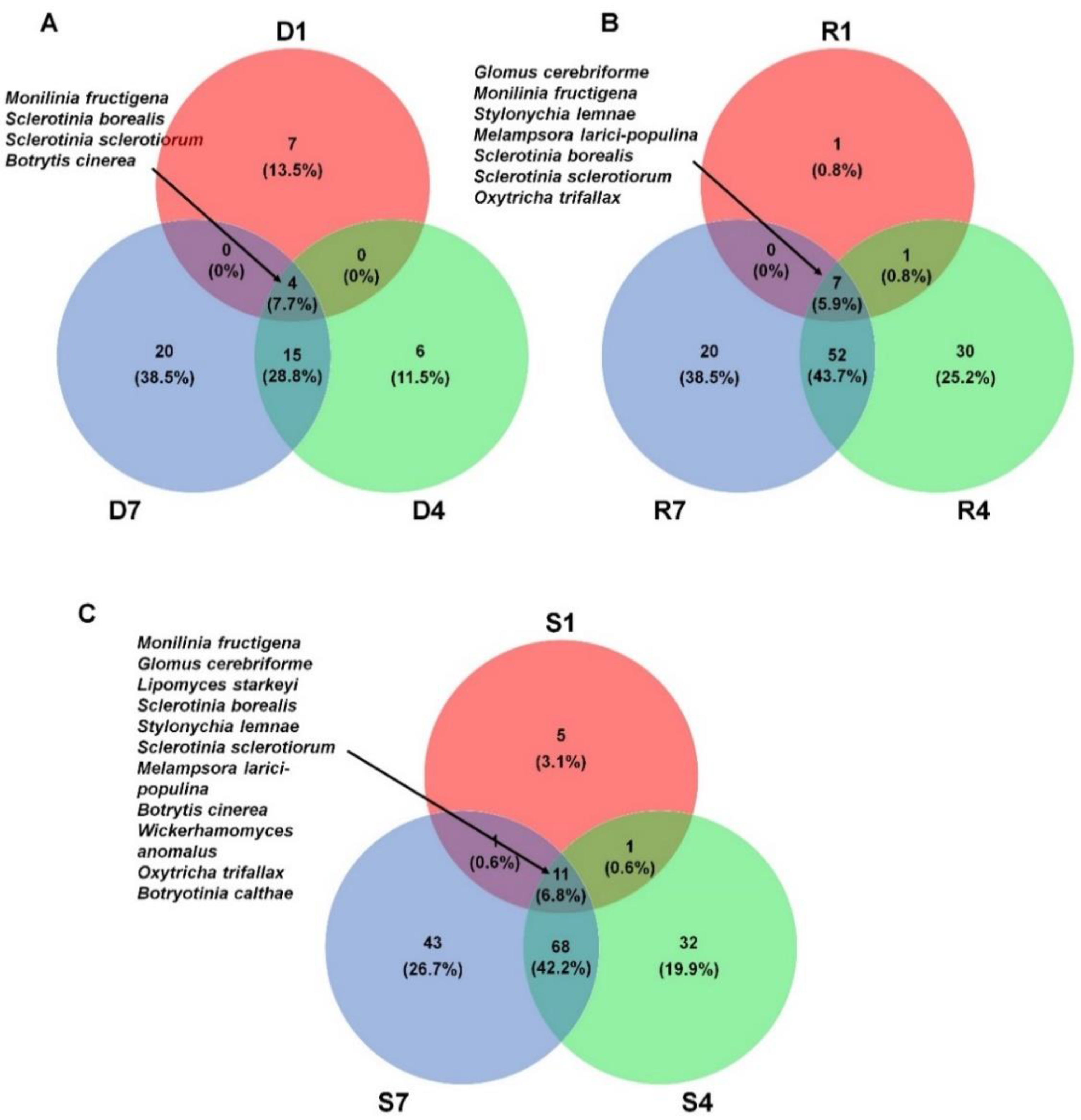
2.6. Comparison of Identified Fungal Species among Different Libraries and Samples
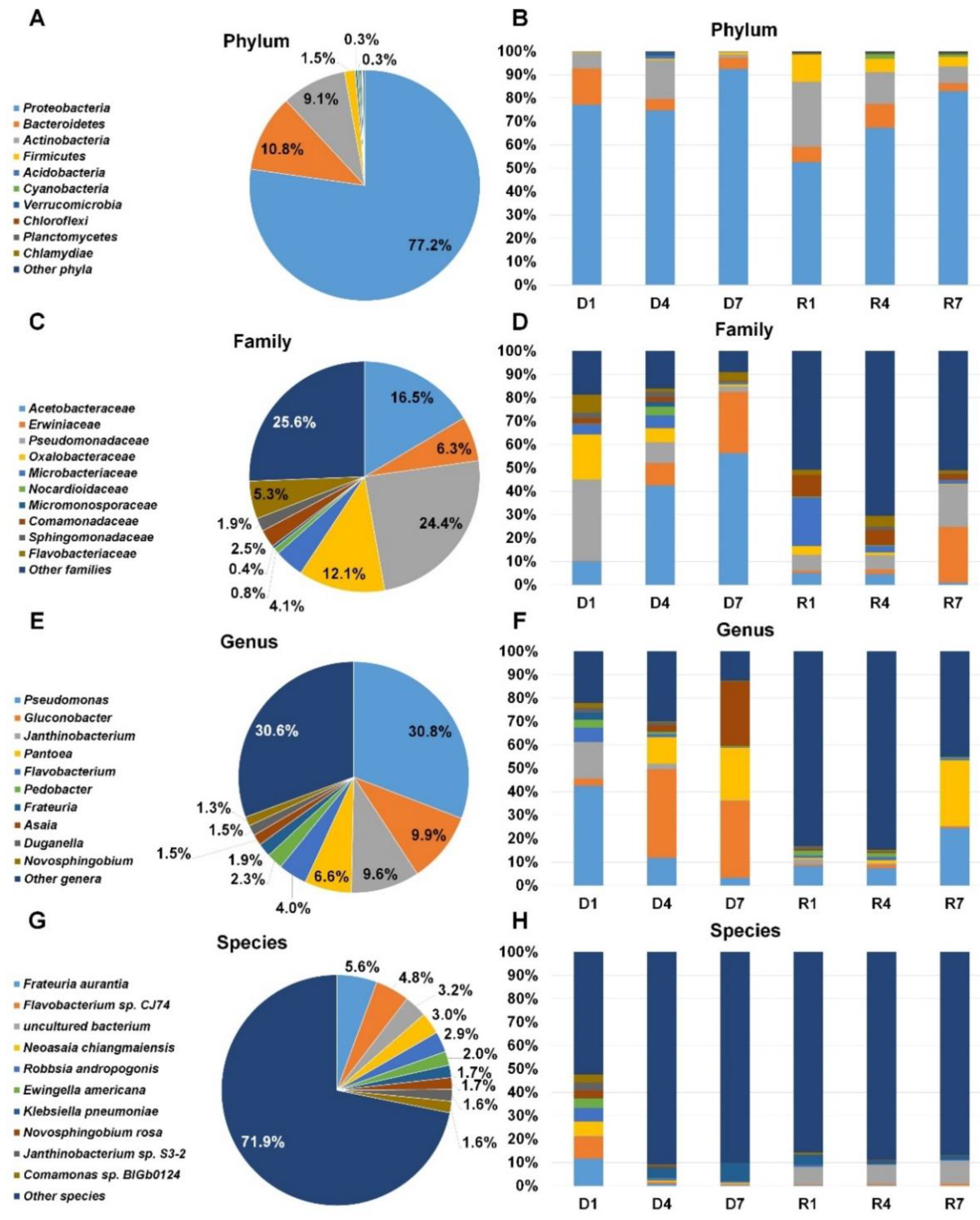
2.7. Classification of Identified Bacteria According to Bacterial Taxonomy
2.8. Identification of Viruses from Six Libraries
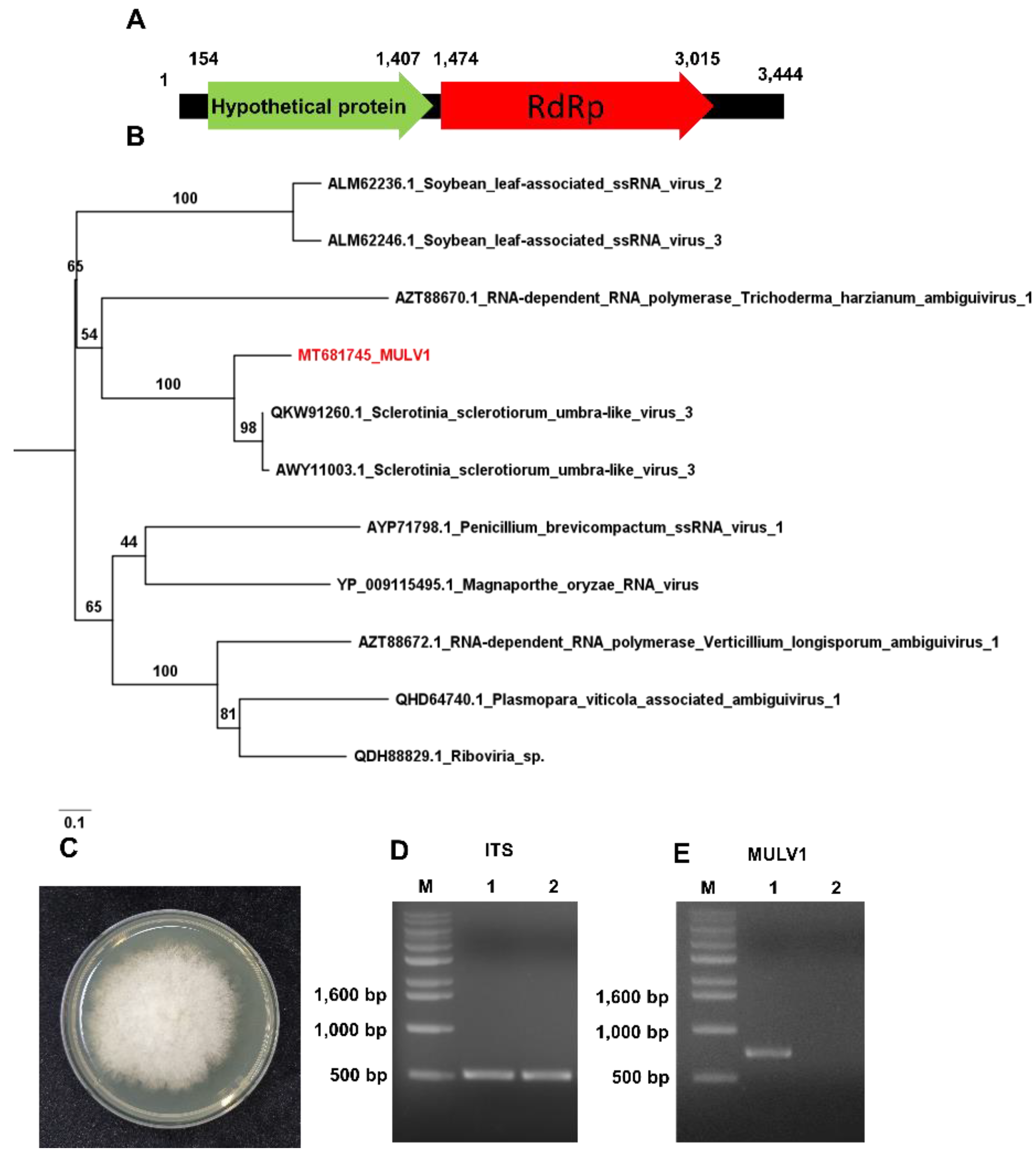
2.9. Identification of Plant Viruses, Viroids and a Novel Mycovirus
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Extraction of Nucleic Acids from the Mummified Peach Fruits
4.3. Library Preparation and Next-Generation Sequencing
4.4. Bioinformatic Analyses
4.5. De Novo Transcriptome Assembly and Virus Identification
4.6. RT-PCR and Sanger-Sequencing
4.7. Construction of Phylogenetic Tree for the Novel Virus
4.8. Confirmation of the Host for the Novel Mycovirus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, S.; Zheng, Q.; Xu, B.; Liu, J. Identification of the fungal pathogens of postharvest disease on peach fruits and the control mechanisms of Bacillus subtilis JK-14. Toxins 2019, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Bernat, M.; Segarra, J.; Xu, X.-M.; Casals, C.; Usall, J. Influence of temperature on decay, mycelium development and sporodochia production caused by Monilinia fructicola and M. laxa on stone fruits. Food Microbiol. 2017, 64, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, O.A.; Baykal, N. Evaluation of the use of microwave power for the control of postharvest diseases of peaches. Postharvest Biol. Technol. 2002, 26, 237–240. [Google Scholar] [CrossRef]
- Inoue, K.; Hideo, N. Black spot of peach caused by Alternaria alternata (Fr.) Keissler. J. Gen. Plant Pathol. 2000, 66, 18–22. [Google Scholar] [CrossRef]
- Thomidis, T.; Michailides, T.; Exadaktylou, E. Contribution of pathogens to peach fruit Rot in northern Greece and their sensitivity to iprodione, carbendazim, thiophanate-methyl and tebuconazole fungicides. J. Phytopathol. 2009, 157, 194–200. [Google Scholar] [CrossRef]
- De Cal, A.; Melgarejo, P. Interactions of pesticides and mycoflora of peach twigs. Mycol. Res. 1992, 96, 1105–1113. [Google Scholar] [CrossRef]
- Yang, Z.; Cao, S.; Cai, Y.; Zheng, Y. Combination of salicylic acid and ultrasound to control postharvest blue mold caused by Penicillium expansum in peach fruit. Innov. Food Sci. Emerg. Technol. 2011, 12, 310–314. [Google Scholar] [CrossRef]
- Manici, L.M.; Caputo, F. Soil fungal communities as indicators for replanting new peach orchards in intensively cultivated areas. Eur. J. Agron. 2010, 33, 188–196. [Google Scholar] [CrossRef]
- Obi, V.I.; Barriuso, J.J.; Gogorcena, Y. Peach brown rot: Still in search of an ideal management option. Agriculture 2018, 8, 125. [Google Scholar] [CrossRef]
- Pancher, M.; Ceol, M.; Corneo, P.E.; Longa, C.M.O.; Yousaf, S.; Pertot, I.; Campisano, A. Fungal endophytic communities in grapevines (Vitis vinifera L.) respond to crop management. Appl. Environ. Microbiol. 2012, 78, 4308–4317. [Google Scholar] [CrossRef]
- Evans, T.N.; Seviour, R.J. Estimating biodiversity of fungi in activated sludge communities using culture-independent methods. Microb. Ecol. 2012, 63, 773–786. [Google Scholar] [CrossRef] [PubMed]
- McCartney, H.A.; Foster, S.J.; Fraaije, B.A.; Ward, E. Molecular diagnostics for fungal plant pathogens. Pest Manag. Sci. Former. Pestic. Sci. 2003, 59, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Schaad, N.W.; Frederick, R.D. Real-time PCR and its application for rapid plant disease diagnostics. Can. J. Plant Pathol. 2002, 24, 250–258. [Google Scholar] [CrossRef]
- Gell, I.; Cubero, J.; Melgarejo, P. Two different PCR approaches for universal diagnosis of brown rot and identification of Monilinia spp. in stone fruit trees. J. Appl. Microbiol. 2007, 103, 2629–2637. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Mu, F.; Xie, J.; Cheng, S.; You, M.P.; Barbetti, M.J.; Jia, J.; Wang, Q.; Cheng, J.; Fu, Y.; Chen, T. Virome characterization of a collection of S. sclerotiorum from Australia. Front. Microbiol. 2018, 8, 2540. [Google Scholar] [CrossRef]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Garrido-Sanz, L.; Senar, M.À.; Piñol, J. Estimation of the relative abundance of species in artificial mixtures of insects using low-coverage shotgun metagenomics. Metabarcoding Metagenom. 2020, 4, e48281. [Google Scholar] [CrossRef]
- Grzegorczyk, M.; Żarowska, B.; Restuccia, C.; Cirvilleri, G. Postharvest biocontrol ability of killer yeasts against Monilinia fructigena and Monilinia fructicola on stone fruit. Food Microbiol. 2017, 61, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, I.; Bassi, D.; Eduardo, I.; Ciacciulli, A.; Pirona, R.; Rossini, L.; Vecchietti, A. QTL mapping for brown rot (Monilinia fructigena) resistance in an intraspecific peach (Prunus persica L. Batsch) F1 progeny. Tree Genet. Genomes 2014, 10, 1223–1242. [Google Scholar] [CrossRef]
- Oh, H.-T.; Choi, I.-Y.; Kim, J.; Na, Y.-E.; Lee, W.-H.; Lee, K.-J.; Shin, H.-D. Characteristics of Brown Rot Caused by Monilinia fructicola on Stone Fruit in Korea. Res. Plant Dis. 2017, 23, 322–333. [Google Scholar]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Di Rienzi, S.C.; Poole, A.C.; Koren, O.; Walters, W.A.; Caporaso, J.G.; Knight, R.; Ley, R.E. Conducting a microbiome study. Cell 2014, 158, 250–262. [Google Scholar] [CrossRef]
- Landi, L.; Angelini, R.M.D.M.; Pollastro, S.; Abate, D.; Faretra, F.; Romanazzi, G. Genome sequence of the brown rot fungal pathogen Monilinia fructigena. BMC Res. Notes 2018, 11, 1–3. [Google Scholar] [CrossRef]
- De Miccolis Angelini, R.M.; Romanazzi, G.; Pollastro, S.; Rotolo, C.; Faretra, F.; Landi, L. New high-quality draft genome of the brown rot fungal pathogen Monilinia fructicola. Genome Biol. Evol. 2019, 11, 2850–2855. [Google Scholar] [CrossRef]
- Rivera, Y.; Zeller, K.; Srivastava, S.; Sutherland, J.; Galvez, M.; Nakhla, M.; Poniatowska, A.; Schnabel, G.; Sundin, G.; Abad, Z.G. Draft genome resources for the phytopathogenic fungi Monilinia fructicola, M. fructigena, M. polystroma, and M. laxa, the causal agents of brown rot. Phytopathology 2018, 108, 1141–1142. [Google Scholar] [CrossRef]
- Angelini, R.M.D.M.; Abate, D.; Rotolo, C.; Gerin, D.; Pollastro, S.; Faretra, F. De novo assembly and comparative transcriptome analysis of Monilinia fructicola, Monilinia laxa and Monilinia fructigena, the causal agents of brown rot on stone fruits. BMC Genom. 2018, 19, 436. [Google Scholar]
- Ioos, R.; Frey, P. Genomic variation within Monilinia laxa, M. fructigena and M. fructicola, and application to species identification by PCR. Eur. J. Plant Pathol. 2000, 106, 373–378. [Google Scholar] [CrossRef]
- Côté, M.-J.; Tardif, M.-C.; Meldrum, A. Identification of Monilinia fructigena, M. fructicola, M. laxa, and Monilia polystroma on inoculated and naturally infected fruit using multiplex PCR. Plant Dis. 2004, 88, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Donovan, P.D.; Gonzalez, G.; Higgins, D.G.; Butler, G.; Ito, K. Identification of fungi in shotgun metagenomics datasets. PLoS ONE 2018, 13, e0192898. [Google Scholar] [CrossRef] [PubMed]
- Keepers, K.G.; Pogoda, C.S.; White, K.H.; Anderson Stewart, C.R.; Hoffman, J.M.; Ruiz, A.M.; McCain, C.M.; Lendemer, J.C.; Kane, N.C.; Tripp, E.A. Whole genome shotgun sequencing detects greater lichen fungal diversity than amplicon-based methods in environmental samples. Front. Ecol. Evol. 2019, 7, 484. [Google Scholar] [CrossRef]
- Jo, Y.; Lian, S.; Chu, H.; Cho, J.K.; Yoo, S.-H.; Choi, H.; Yoon, J.-Y.; Choi, S.-K.; Lee, B.C.; Cho, W.K. Peach RNA viromes in six different peach cultivars. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Huo, Y.; Shen, J.; Wu, H.; Zhang, C.; Guo, L.; Yang, J.; Li, W. Widespread 3′-end uridylation in eukaryotic RNA viruses. Sci. Rep. 2016, 6, 25454. [Google Scholar] [CrossRef]
- Kuske, C.R.; Hesse, C.N.; Challacombe, J.F.; Cullen, D.; Herr, J.R.; Mueller, R.C.; Tsang, A.; Vilgalys, R. Prospects and challenges for fungal metatranscriptomics of complex communities. Fungal Ecol. 2015, 14, 133–137. [Google Scholar] [CrossRef]
- Choma, M.; Bárta, J.; Šantrůčková, H.; Urich, T. Low abundance of Archaeorhizomycetes among fungi in soil metatranscriptomes. Sci. Rep. 2016, 6, 38455. [Google Scholar] [CrossRef]
- Cottier, F.; Srinivasan, K.G.; Yurieva, M.; Liao, W.; Poidinger, M.; Zolezzi, F.; Pavelka, N. Advantages of meta-total RNA sequencing (MeTRS) over shotgun metagenomics and amplicon-based sequencing in the profiling of complex microbial communities. NPJ Biofilms Microbiomes 2018, 4, 1–7. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R4 | R7 | ||||||
|---|---|---|---|---|---|---|---|---|
| Virus | Accession No. | Size | Coverage | Reads | Coverage | Reads | Coverage | Reads |
| Apple chlorotic leaf spot virus | NC_001409.1 | 7555 | 0 | 0 | 0.3273 | 36 | 0 | 0 |
| Hop stunt viroid | NC_001351.1 | 302 | 0.5364 | 2 | 19.3444 | 68 | 0 | 0 |
| Peach latent mosaic viroid | NC_003636.1 | 337 | 7.5905 | 30 | 8.095 | 35 | 1.9822 | 8 |
| Asian prunus virus 2 | NC_028868.1 | 9362 | 0.0092 | 1 | 0.9509 | 110 | 0 | 0 |
| Asian prunus virus 3 | NC_028975.1 | 9654 | 0 | 0 | 0.1859 | 28 | 0 | 0 |
| Monilinia umbra-like virus | MT681745 | 4147 | 29.8676 | 1272 | 117.3161 | 4904 | 0.2358 | 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, Y.; Back, C.-G.; Choi, H.; Cho, W.K. Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics. Plants 2020, 9, 1052. https://doi.org/10.3390/plants9081052
Jo Y, Back C-G, Choi H, Cho WK. Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics. Plants. 2020; 9(8):1052. https://doi.org/10.3390/plants9081052
Chicago/Turabian StyleJo, Yeonhwa, Chang-Gi Back, Hoseong Choi, and Won Kyong Cho. 2020. "Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics" Plants 9, no. 8: 1052. https://doi.org/10.3390/plants9081052
APA StyleJo, Y., Back, C.-G., Choi, H., & Cho, W. K. (2020). Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics. Plants, 9(8), 1052. https://doi.org/10.3390/plants9081052