MYO, a Candidate Gene for Haploid Induction in Maize Causes Male Sterility


Abstract
1. Introduction
2. Results
2.1. Sequence Alignment
2.2. Silencing Efficiency
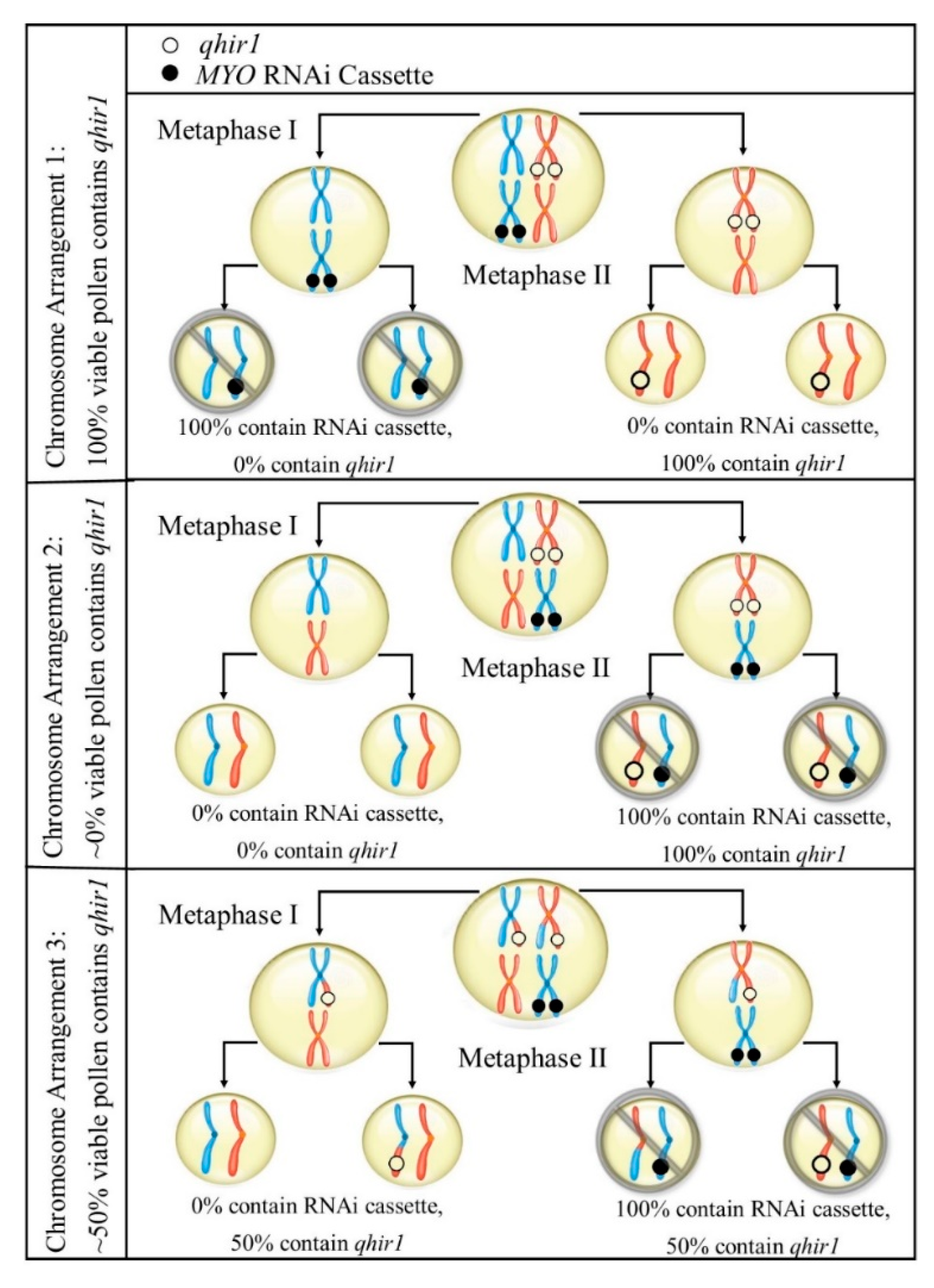
2.3. Haploid Induction Rate
2.4. Male Fertility
3. Discussion
4. Materials and Methods
4.1. Identification of Gene of Interest
4.2. Production of Transgenic Materials
4.3. Development of Control Lines
4.4. Development of Transgenic Families
4.5. Test Crossing and Evaluation of HIR
4.6. Experimental Design and Statistical Analysis for HIR
4.7. Male Fertility Analysis
4.8. Silencing Efficiency by Quantitative PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prigge, V.; Xu, X.W.; Li, L.; Babu, R.; Chen, S.; Atlin, G.N.; Melchinger, A.E. New insights into the genetics of in vivo induction of maternal haploids, the backbone of doubled haploid technology in maize. Genetics 2012, 190, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Ren, J.; Mei, M.; Frei, U.K.; Trampe, B.; Lübberstedt, T.; Goldman, I.L.; Ortiz, R. Maize doubled haploids. In Plant Breeding Reviews; Janick, J., Ed.; Wiley Blackwell: Hoboken, NJ, USA, 2016; Volume 40, pp. 123–160. [Google Scholar]
- Deimling, S.; Röber, F.K.; Geiger, H.H. Methodology and genetics of in vivo haploid induction in maize. Vortr. Pflanzenzüchtg 1997, 38, 203–224. [Google Scholar]
- Barret, P.; Brinkmann, M.; Beckert, M. A major locus expressed in the male gametophyte with incomplete penetrance is responsible for in situ gynogenesis in maize. Theor. Appl. Genet. 2008, 117, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, W.; Zhong, Y.; Dong, X.; Hu, H.; Tian, X.; Wang, L.; Chen, B.; Chen, C.; Melchinger, A.E.; et al. Fine mapping of qhir8 affecting in vivo haploid induction in maize. Theor. Appl. Genet. 2015, 128, 2507–2515. [Google Scholar] [CrossRef]
- Hu, H.; Schrag, T.A.; Peis, R.; Unterseer, S.; Schipprack, W.; Chen, S.; Lai, J.; Yan, J.; Prasanna, B.M.; Nair, S.; et al. The Genetic Basis of Haploid Induction in Maize Identified with a Novel Genome-Wide Association Method. Genetics 2016, 202, 1267–1276. [Google Scholar] [CrossRef]
- Dong, X.; Xu, X.; Miao, J.; Li, L.; Zhang, D.; Mi, X.; Liu, C.; Tian, X.; Melchinger, A.E.; Chen, S.; et al. Fine mapping of qhir1 influencing in vivo haploid induction in maize. Theor. Appl. Genet. 2013, 126, 1713–1720. [Google Scholar] [CrossRef]
- Kelliher, T.; Starr, D.; Richbourg, L.; Chintamanani, S.; Delzer, B.; Nuccio, M.L.; Green, J.; Chen, Z.; McCuiston, J.; Wang, W.; et al. Matrilineal, a sperm-specific phospholipase, triggers maize haploid induction. Nature 2017, 542, 105–109. [Google Scholar] [CrossRef]
- Gilles, L.; Khaled, A.; Laffaire, J.B.; Chaignon, S.; Gendrot, G.; Laplaige, J.; Bergès, H.; Beydon, G.; Bayle, V.; Barret, P.; et al. Loss of pollen-specific phospholipase NOT LIKE DAD triggers gynogenesis in maize. EMBO J. 2017, 36, 707–717. [Google Scholar] [CrossRef]
- Liu, C.X.; Li, X.; Meng, D.X.; Zhong, Y.; Chen, C.; Dong, X.; Xu, X.; Chen, B.; Li, W.; Li, L.; et al. A 4bp insertion at ZmPLA1 encoding a putative phospholipase A generates haploid induction in maize. Mol. Plant 2017, 10, 520–522. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, C.; Qi, X.; Jiao, Y.; Wang, D.; Wang, Y.; Liu, Z.; Chen, C.; Chen, B.; Tian, X.; et al. Mutation of ZmDMP enhances haploid induction in maize. Nat. Plants 2019, 5, 575–580. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; I Hurwitz, D.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Peremyslov, V.V.; Morgun, E.A.; Kurth, E.G.; Makarova, K.S.; Koonin, E.V.; Dolja, V.V. Identification of myosin XI receptors in Arabidopsis defines a distinct class of transport vesicles. Plant Cell 2013, 25, 3022–3038. [Google Scholar] [CrossRef] [PubMed]
- Shimmen, T. The sliding theory of cytoplasmic streaming: Fifty years of progress. J. Plant Res. 2007, 120, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar]
- Trentin, H.U. Study and Improvement of Maize Maternal Haploid Inducers (Doctoral Dissertation); Iowa State University: Ames, IA, USA, 2019. [Google Scholar]
- Kinkema, M.; Wang, H.; Schiefelbein, J. Molecular analysis of the myosin gene family in Arabidopsis thaliana. Plant Mol. Biol. 1994, 26, 1139–1153. [Google Scholar] [CrossRef]
- Sekhon, R.S.; Lin, H.; Childs, K.L.; Hansey, C.N.; Buell, C.R.; De Leon, N.; Kaeppler, S.M. Genome-wide atlas of transcription during maize development. Plant J. 2011, 66, 553–563. [Google Scholar] [CrossRef]
- Stelpflug, S.C.; Sekhon, R.S.; Vaillancourt, B.; Hirsch, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. An expanded maize gene expression atlas based on RNA-sequencing and its use to. Plant Genome 2015, 1–16. [Google Scholar] [CrossRef]
- Röber, F.K.; Gordillo, G.A.; Geiger, H.H. In vivo haploid induction in maize-performance of new inducers and significance of doubled haploid lines in hybrid breeding. Maydica 2005, 50, 275–283. [Google Scholar]
- Dai, X.; Zhao, P.X. pssRNAMiner: A plant short small RNA regulatory cascade analysis server. Nucleic Acids Res. 2008, 36, W114–W118. [Google Scholar] [CrossRef]
- Frame, B.; Main, M.; Schick, R.; Wang, K. Genetic transformation using maize immature zygotic embryos. In Plant Embryo Culture: Methods and Protocols. Methods in Molecular Biology; Thorpe, A., Yeung, E.C., Eds.; Springer Science: New York, NY, USA, 2011; pp. 327–341. [Google Scholar]
- Russel, W.A. Crop Science. Madison, WI. Crop Sci. 1972, 12, 721. [Google Scholar]
- Chase, S.S. Monoploid frequencies in a commercial double cross hybrid maize, and in its component single cross hybrids and inbred lines. Genetics 1949, 34, 328–332. [Google Scholar] [PubMed]
- Chase, S.S. Monoploids in maize. In Heterosis; Gowen, J.W., Ed.; Iowa State College Press: Ames, IA, USA, 1952; pp. 389–399. [Google Scholar]
- Pukelsheim, F. The Three Sigma Rule. Am. Stat. 2012, 48, 88–91. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the Double Delta CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Duncan, D.; Morrow, D.J.; Fernandes, J.; Walbot, V. Transcriptome profiling of maize anthers using genetic ablation to analyze pre-meiotic and tapetal cell types. Plant J. 2007, 50, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Manoli, A.; Sturaro, A.; Trevisan, S.; Quaggiotti, S.; Nonis, A. Evaluation of candidate reference genes for qPCR in maize. J. Plant Phys. 2012, 169, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | p-Values | Normalized Expression (ΔCT) | Calibrated Expression (ΔΔCT) | Relative Expression (Fold Change) |
|---|---|---|---|---|
| MYO16-1 | 0.04 * | 1.17 | −2.35 | 5.09 |
| MYO18-1 | 0.05 * | 1.80 | −1.72 | 3.30 |
| MYO18-2 | 0.03 * | 1.38 | −2.14 | 4.40 |
| MYO18-3 | 0.03 * | 1.34 | −2.18 | 4.52 |
| MYO18-4 | 0.16 | 2.33 | −1.19 | 2.29 |
| Viking | ------- | 3.52 | 0.00 | 1.00 |
| Group | Cross ID | p-Value | HIR (%) | S.E. |
|---|---|---|---|---|
| Controls | Spontaneous | 0.1000 | ||
| qhir8B73 | 0.0007 ** | 0.2398 | 0.0734 | |
| qhir1,qhir8B73 | 0.0016 ** | 1.157 | 0.1708 | |
| MYO16-1 | MYOBC1F1 | 0.0336 * | 0.0829 | 0.0852 |
| qhir1BC2F1 | 0.0918 | 1.120 | 0.3356 | |
| qhir1,MYOBC2F1 | 0.0291 * | 1.250 | 0.3449 | |
| MYO18-1 | MYOBC1F1 | 0.1723 | 0.2333 | 0.1735 |
| qhir1BC2F1 | 0.5274 | 0.8517 | 0.3650 | |
| qhir1,MYOBC2F1 | 0.0030 ** | 1.581 | 0.4294 | |
| MYO18-2 | MYOBC1F1 | 0.0958 | 0.1207 | 0.1245 |
| qhir1BC2F1 | 0.1120 | 1.216 | 0.4717 | |
| qhir1,MYO BC2F1 | 0.0001 ** | 2.015 | 0.5555 | |
| MYO18-3 | MYOBC1F1 | 0.1893 | 0.2413 | 0.1794 |
| qhir1 BC2F1 | 0.9402 | 0.6711 | 0.3063 | |
| qhir1,MYO BC2F1 | 0.0429 * | 1.254 | 0.3681 |
| Male Parent | Survival Rate (%) | S.E. | Effect | p-Values |
|---|---|---|---|---|
| B73 | 47.45 | 1.16 | Event | 0.0001 |
| MYO16-1 | 0 | 1.77 | Female | <0.0001 |
| MYO18-1 | 12.42 | 1.77 | Event * Female | 0.0001 |
| MYO18-2 | 20.11 | 2.97 | ||
| MYO18-3 | 8.48 | 1.77 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanous, K.; Lübberstedt, T.; Ibrahim, R.; Frei, U.K. MYO, a Candidate Gene for Haploid Induction in Maize Causes Male Sterility. Plants 2020, 9, 773. https://doi.org/10.3390/plants9060773
Vanous K, Lübberstedt T, Ibrahim R, Frei UK. MYO, a Candidate Gene for Haploid Induction in Maize Causes Male Sterility. Plants. 2020; 9(6):773. https://doi.org/10.3390/plants9060773
Chicago/Turabian StyleVanous, Kimberly, Thomas Lübberstedt, Rania Ibrahim, and Ursula K. Frei. 2020. "MYO, a Candidate Gene for Haploid Induction in Maize Causes Male Sterility" Plants 9, no. 6: 773. https://doi.org/10.3390/plants9060773
APA StyleVanous, K., Lübberstedt, T., Ibrahim, R., & Frei, U. K. (2020). MYO, a Candidate Gene for Haploid Induction in Maize Causes Male Sterility. Plants, 9(6), 773. https://doi.org/10.3390/plants9060773