Characterization of Gene Isoforms Related to Cellulose and Lignin Biosynthesis in Kenaf (Hibiscus cannabinus L.) Mutant

 ,
,  ,
, 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Transcriptome Assembly and Gene Annotation
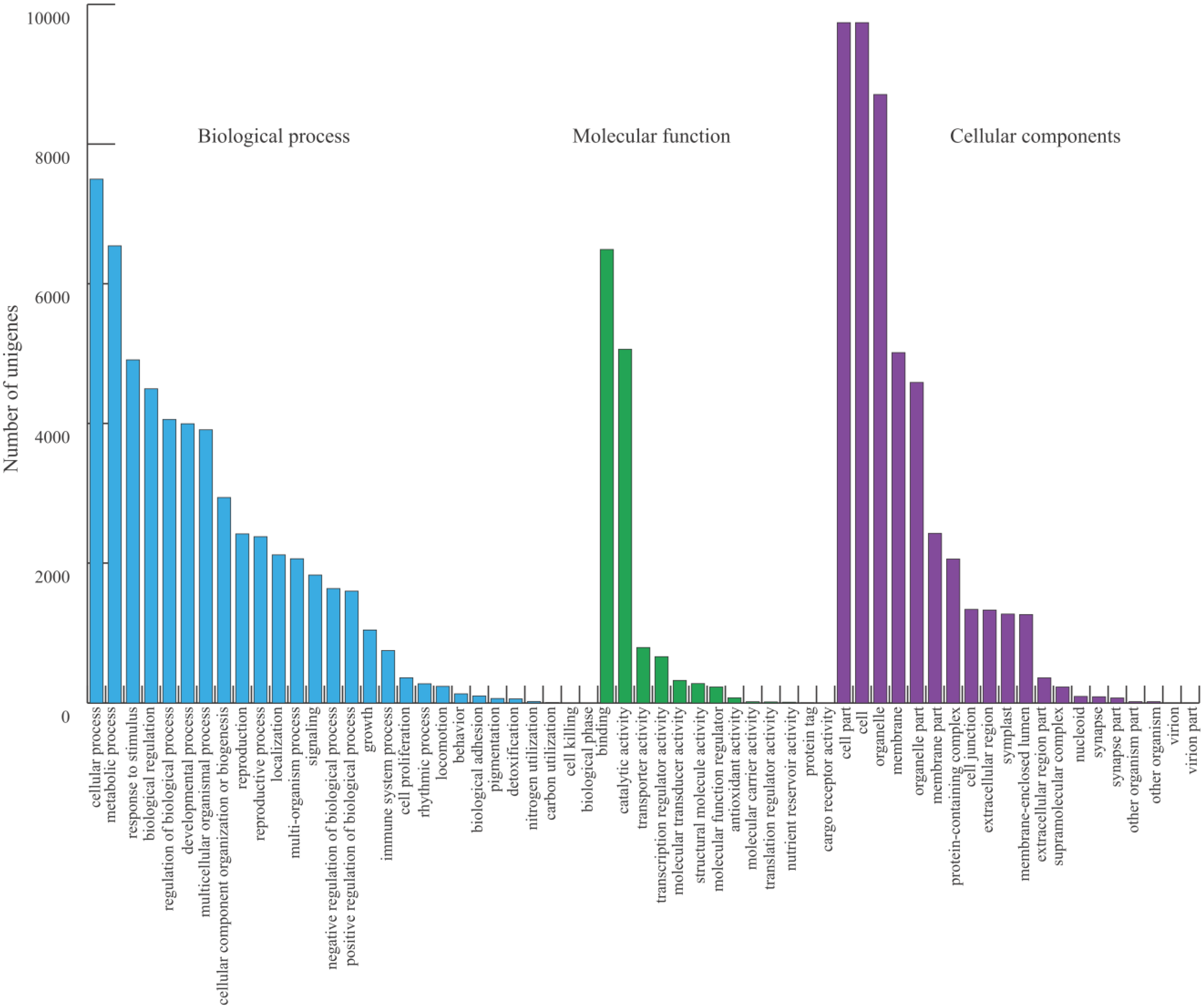
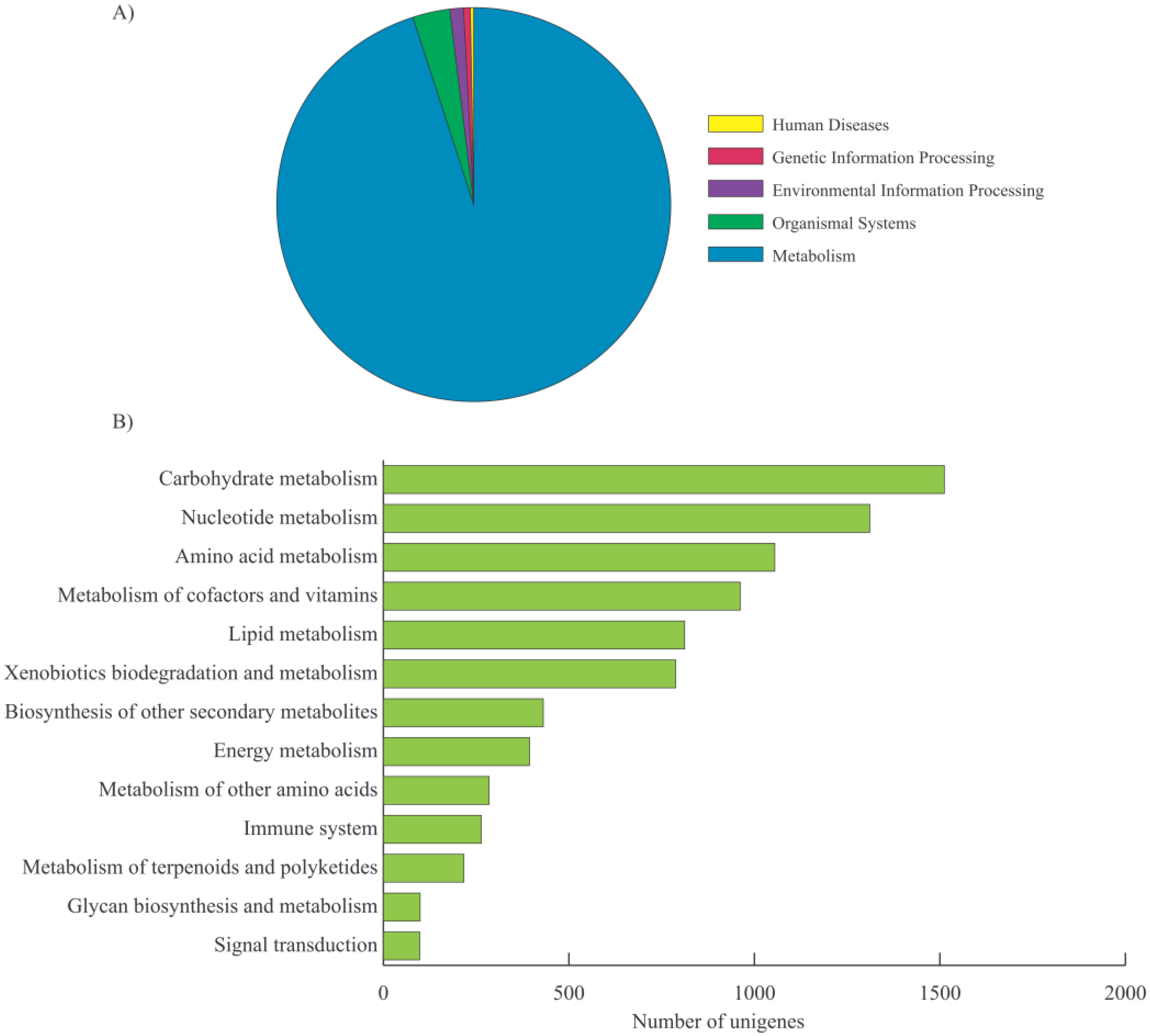
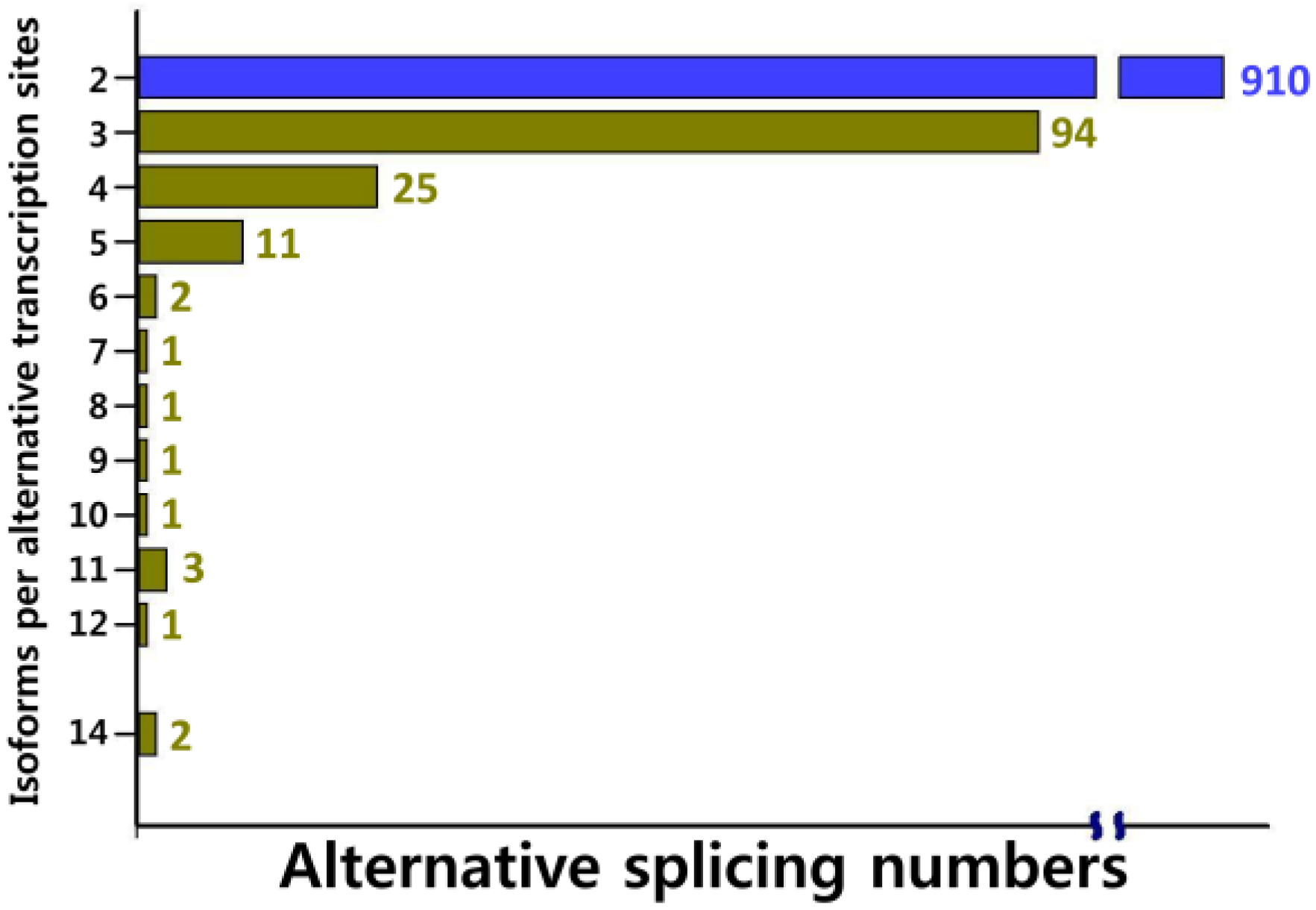
2.2. Functional Annotation, KEGG Classification, and Isoform Analysis
2.3. Identification of Major Genes Involved in Cellulose and Lignin Biosynthesis
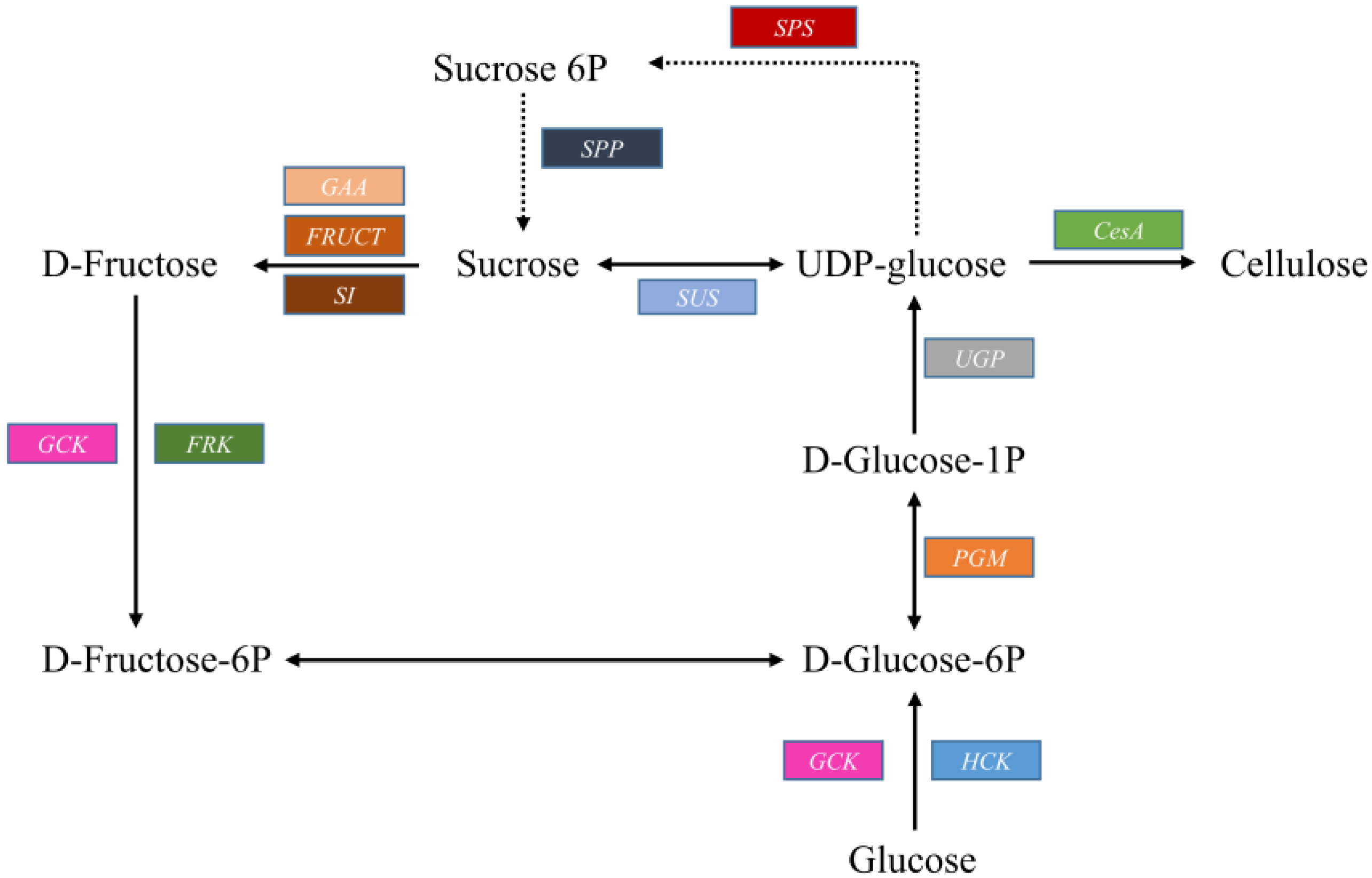
2.3.1. Cellulose Biosynthesis
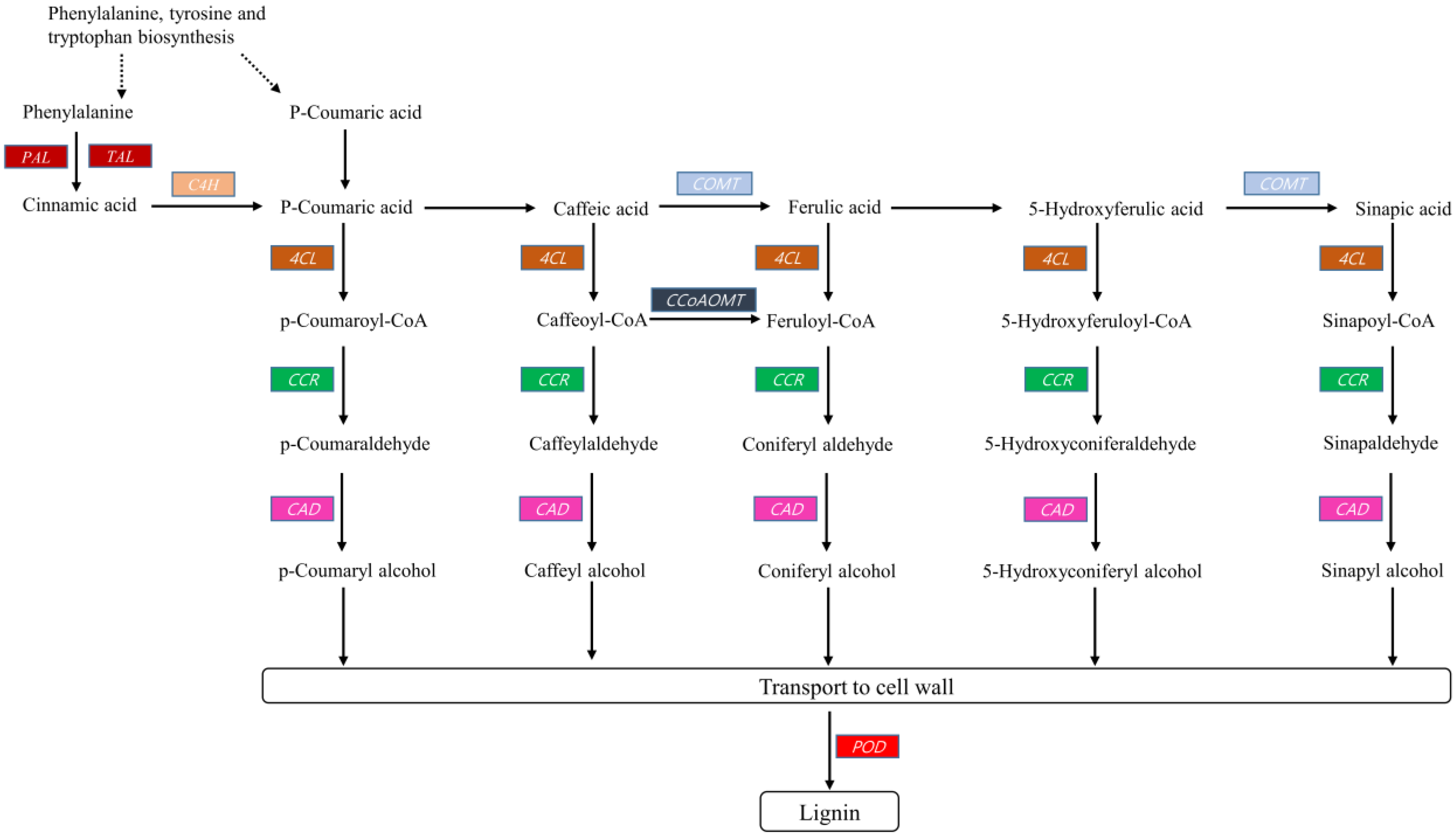
2.3.2. Lignin Biosynthesis
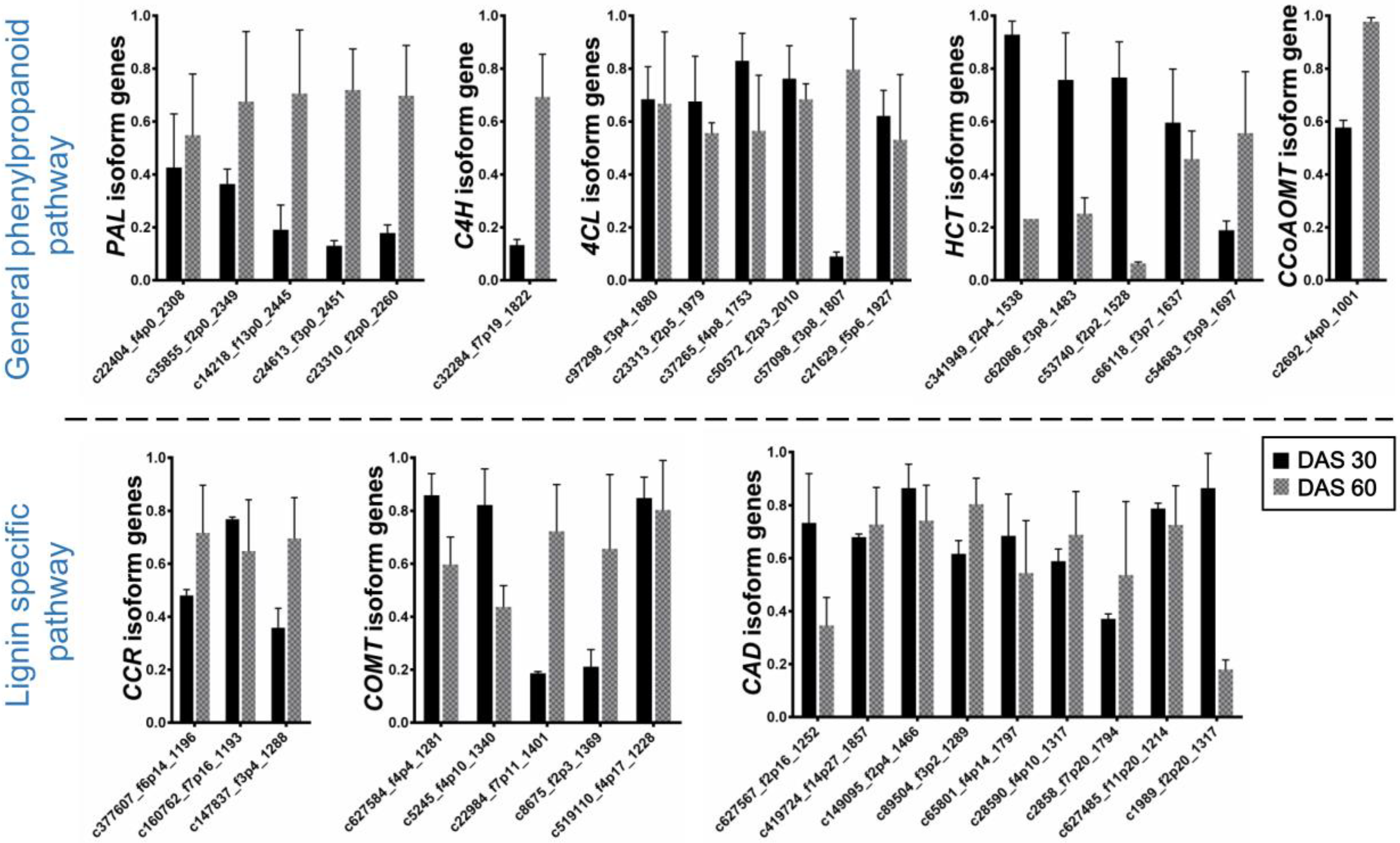
2.4. Expression Analysis of Lignin Biosynthesis Genes
2.4.1. General Phenylpropanoid Pathway
2.4.2. Lignin Specific Pathway
3. Discussion
4. Materials and Methods
4.1. Plant Material and RNA Extraction
4.2. PacBio SMRT Iso-Seq Sequencing and Data Analysis
4.3. Functional Annotation and Classification
4.4. Isoform Grouping
4.5. Quantitative PCR/Expression Analysis of Lignin Biosynthesis Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexopoulou, E.; Papatheohari, Y.; Christou, M.; Monti, A. Origin, Description, Importance, and Cultivation Area of Kenaf. In Kenaf: A Multi-Purpose Crop for Several Industrial Applications: New insights from the Biokenaf Project; Monti, A., Alexopoulou, E., Eds.; Springer London: London, UK, 2013; Volume 1, pp. 1–15. [Google Scholar]
- Dempsey, J.M. Fiber Crops; Univ. Presses of Florida: Gainesville, FL, USA, 1975; p. 470. [Google Scholar]
- Bowyer, J.L.; Stockmann, V.E. Agricultural residues: An exciting bio-based raw material for the global panels industry. Forest Prod. J 2001, 51, 10–21. [Google Scholar]
- Bañuelos, G.S.; Bryla, D.; Cook, C.G. Vegetative production of kenaf and canola under irrigation in central California. Ind. Crop. Prod. 2002, 15, 237–245. [Google Scholar] [CrossRef]
- Lam, T.B.T.; Hori, K.; Iiyama, K. Structural characteristics of cell walls of kenaf (Hibiscus cannabinus L.) and fixation of carbon dioxide. J. Wood Sci. 2003, 49, 255–261. [Google Scholar] [CrossRef]
- Ryu, J.; Kwon, S.-J.; Ahn, J.-W.; Ha, B.-K.; Jeong, S.W.; Bin Im, S.; Kim, J.-B.; Young-Keun, S.H.K.; Kang, S.-Y. Evaluation of Nutritive Value and Identification of Fungi in Silage from New Kenaf (Hibiscus cannabinus) Cultivars. Int. J. Agric. Boil. 2016, 18, 1159–1168. [Google Scholar] [CrossRef]
- Webber, C.L.; Bledsoe, V.K. Plant maturity and kenaf yield components. Ind. Crop. Prod. 2002, 16, 81–88. [Google Scholar] [CrossRef]
- Kang, S.-Y.; Kwon, S.-J.; Jeong, S.W.; Kim, J.-B.; Kim, S.H.; Ryu, J. An Improved Kenaf Cultivar ‘Jangdae’ with Seed Harvesting in Korea. Korean J. Breed. Sci. 2016, 48, 349–354. [Google Scholar] [CrossRef]
- Hrdlicková, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2016, 8, e1364. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2017, 217, 163–178. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, X.; Zhao, X.; Liu, C.; Wang, C.; Zhang, Z.; Zhang, C.; Wei, Q.; Wang, Q.; Yan, H.; et al. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.). Genomics 2011, 98, 47–55. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Tao, A.; Fang, P.; Qi, J. Development and Characterization of 1,906 EST-SSR Markers from Unigenes in Jute (Corchorus spp.). PLoS ONE 2015, 10, e0140861. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, X.; Xu, J.; Lin, L.; Qi, J. De novo assembly of kenaf (Hibiscus cannabinus) transcriptome using Illumina sequencing for gene discovery and marker identification. Mol. Breed. 2015, 35, 192. [Google Scholar] [CrossRef]
- Chen, P.; Ran, S.; Li, R.; Huang, Z.; Qian, J.; Yu, M.; Zhou, R. Transcriptome de novo assembly and differentially expressed genes related to cytoplasmic male sterility in kenaf (Hibiscus cannabinus L.). Mol. Breed. 2014, 34, 1879–1891. [Google Scholar] [CrossRef]
- Lyu, J.I.; Choi, H.-I.; Ryu, J.; Kwon, S.-J.; Jo, Y.D.; Hong, M.J.; Kim, J.-B.; Ahn, J.-W.; Kang, S.-Y. Transcriptome Analysis and Identification of Genes Related to Biosynthesis of Anthocyanins and Kaempferitrin in Kenaf (Hibiscus cannabinus L.). J. Plant Boil. 2020, 63, 51–62. [Google Scholar] [CrossRef]
- Baker, M. De novo genome assembly: What every biologist should know. Nat. Methods 2012, 9, 333–337. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.C.; Chen, N.; Wang, D.W. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef]
- Guerriero, G.; Fugelstad, J.; Bulone, V. What Do We Really Know about Cellulose Biosynthesis in Higher Plants? J. Integr. Plant Boil. 2010, 52, 161–175. [Google Scholar] [CrossRef]
- Kimura, S.; Kondo, T. Recent progress in cellulose biosynthesis. J. Plant Res. 2002, 115, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.A.; Ponnala, L.; Weber, C.A. Strategies for transcriptome analysis in nonmodel plants. Am. J. Bot. 2012, 99, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Jo, I.-H.; Lee, J.; Hong, C.E.; Lee, D.J.; Bae, W.; Park, S.-G.; Ahn, Y.J.; Kim, Y.C.; Kim, J.U.; Lee, J.W.; et al. Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome. Genes 2017, 8, 228. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, J.; Zhao, S.; Hou, Y.; Li, F.; Tai, Y.; Wan, X.; Wei, C. Transcriptome Profiling Using Single-Molecule Direct RNA Sequencing Approach for In-depth Understanding of Genes in Secondary Metabolism Pathways of Camellia sinensis. Front. Plant Sci. 2017, 8, 8. [Google Scholar] [CrossRef]
- Ghosh, R.; Choi, B.; Cho, B.-K.; Lim, H.-S.; Park, S.-U.; Bae, H.-J.; Natarajan, S.; Bae, H. Characterization of Developmental- and Stress-Mediated Expression of Cinnamoyl-CoA Reductase in Kenaf (Hibiscus cannabinus L.). Sci. World J. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Liao, X.; Zhao, Y.; Kong, X.; Khan, A.; Zhou, B.; Liu, N.; Kashif, M.H.; Chen, P.; Wang, H.; Zhou, R. Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 2018, 8, 12714. [Google Scholar] [CrossRef]
- Lips, S.J.; De Heredia, G.M.I.; Kamp, R.G.O.D.; Van Dam, J.E. Water absorption characteristics of kenaf core to use as animal bedding material. Ind. Crop. Prod. 2009, 29, 73–79. [Google Scholar] [CrossRef]
- Nishino, T.; Hirao, K.; Kotera, M.; Nakamae, K.; Inagaki, H. Kenaf reinforced biodegradable composite. Compos. Sci. Technol. 2003, 63, 1281–1286. [Google Scholar] [CrossRef]
- Yu, H.; Yu, C. Study on microbe retting of kenaf fiber. Enzym. Microb. Technol. 2007, 40, 1806–1809. [Google Scholar] [CrossRef]
- Ramawat, K.G.; Ahuja, M.R. Fiber Plants: An Overview. In Fiber Plants: Biology, Biotechnology and Applications; Springer International Publishing: Cham, Switzerland, 2016; Volume 1, pp. 3–15. [Google Scholar]
- Lange, B.M.; Lapierre, C.; Sandermann, H., Jr. Elicitor-Induced Spruce Stress Lignin (Structural Similarity to Early Developmental Lignins). Plant Physiol. 1995, 108, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Choi, H.; An, G. Roles of lignin biosynthesis and regulatory genes in plant development. J. Integr. Plant Boil. 2015, 57, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chapple, C.; Weng, J.-K. Improvement of biomass through lignin modification. Plant J. 2008, 54, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Morrison, W., III; Akin, D.; Archibald, D.; Dodd, R.; Raymer, P.L. Chemical and instrumental characterization of maturing kenaf core and bast. Ind. Crop. Prod. 1999, 10, 21–34. [Google Scholar] [CrossRef]
- Ryu, J.; Kwon, S.-J.; Sung, S.Y.; Kim, W.-J.; Kim, N.S.; Ahn, J.-W.; Kim, J.-B.; Kim, S.H.; Ha, B.-K.; Kang, S.-Y. Molecular cloning, characterization, and expression analysis of lignin biosynthesis genes from kenaf (Hibiscus cannabinus L.). Genes Genom. 2015, 38, 59–67. [Google Scholar] [CrossRef]
- Chiaiese, P.; Ruotolo, G.; Di Matteo, A.; Virzo, A.D.S.; De Marco, A.; Filippone, E. Cloning and expression analysis of kenaf (Hibiscus cannabinus L.) major lignin and cellulose biosynthesis gene sequences and polymer quantification during plant development. Ind. Crop. Prod. 2011, 34, 1072–1078. [Google Scholar] [CrossRef]
- Chowdhury, M.E.K.; Choi, B.; Cho, B.-K.; Kim, J.B.; Park, S.U.; Natarajan, S.; Lim, H.-S.; Bae, H. Regulation of 4CL, encoding 4-coumarate: Coenzyme A ligase, expression in kenaf under diverse stress conditions. Plant Omics 2013, 6, 254–262. [Google Scholar]
- Kim, J.; Choi, B.; Cho, B.-K.; Lim, H.-S.; Kim, J.B.; Natarajan, S.; Kwak, E.; Bae, H. Molecular cloning, characterization and expression of the caffeic acid O-methyltransferase (COMT) ortholog from kenaf (Hibiscus cannabinus). Plant Omics 2013, 6, 246–253. [Google Scholar]
- Kim, M.A.; Rhee, J.-S.; Kim, T.H.; Lee, J.S.; Choi, A.-Y.; Choi, B.-S.; Choi, I.-Y.; Sohn, Y.C. Alternative Splicing Profile and Sex-Preferential Gene Expression in the Female and Male Pacific Abalone Haliotis discus hannai. Genes 2017, 8, 99. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Data | Platform | Number of Sequence |
|---|---|---|---|
| 1 | High quality consensus sequence | RS_IsoSeq | 26,822 |
| 2 | Nonredundant representative sequence | CD-HIT | 19,775 |
| 3 | Reference isoforms | BLASTCLUST and TransDecoder | 15,637 |
| 4 | Final isoforms transcriptome | GMAP and ToFU | 12,694 |
| 5 | Final gene set with representative isoforms | TransDecoder | 11,370 |
| Putative Gene | Enzyme | KEGG Ortholog | Enzyme Code | Unigenes |
|---|---|---|---|---|
| HK | Hexokinase | K00844 | EC:2.7.1.1 | 8 |
| GCK | Glucokinase | K00845 | EC 2.7.1.2 | 4 |
| PGM | Phosphoglucomutase | K01835 | EC 5.4.2.2 | 4 |
| UGP | UDP-glucose pyrophosphorylase | K00963 | EC:2.7.7.9 | 5 |
| CesA | Cellulose synthase | K10999 | EC:2.4.1.12 | 21 |
| SUS | Sucrose synthase | K00695 | EC:2.4.1.13 | 9 |
| SPS | Sucrose-phosphate synthase | K07024 | EC 2.4.1.14 | 5 |
| SPP | Sucrose-6-phosphatase | EC 3.1.3.24 | 5 | |
| GAA | Alpha-glucosidase | K01187 | EC 3.2.1.20 | 19 |
| FRUCT | Beta-fructofuranosidase | K01193 | EC 3.2.1.26 | 11 |
| SI | Sucrase-isomaltase | K01203 | EC 3.2.1.48 | 11 |
| FRK | Fructokinase | K00847 | EC:2.7.1.4 | 7 |
| Putative Gene | Enzyme | KEGG Ortholog | Enzyme Code | Unigenes |
|---|---|---|---|---|
| PAL | Phenylalanine ammonia lyase | K10775 | EC:4.3.1.24 | 5 |
| PAL | Tyrosine ammonia lyase | K13064 | EC:4.3.1.25 | 5 |
| C4H | Cinnamate 4-hydroxylase | K00487 | EC:1.14.13.11 | 1 |
| 4CL | 4-coumarate CoA ligase | K01904 | EC:6.2.1.12 | 6 |
| CCR | Cinnamoyl CoA reductase | K09753 | EC:1.2.1.44 | 3 |
| CAD | Cinnamyl alcohol dehydrogenase | K00083 | EC:1.1.1.195 | 9 |
| HCT | Hydroxycinnamoyl CoA shikimate /quinate phydroxycinnamoyl transferase | K13065 | EC:2.3.1.133 | 5 |
| CCoAOMT | Caffeoyl CoA O-methyltransferase | K00588 | EC:2.1.1.104 | 1 |
| COMT | Caffeic acid O-methyltransferase | K13066 | E2.1.1.68 | 2 |
| POD | Peroxidase | K00430 | EC:1.11.1.7 | 41 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, J.I.; Ramekar, R.; Kim, D.-G.; Kim, J.M.; Lee, M.-K.; Hung, N.N.; Kim, J.-B.; Ahn, J.-W.; Kang, S.-Y.; Choi, I.-Y.; et al. Characterization of Gene Isoforms Related to Cellulose and Lignin Biosynthesis in Kenaf (Hibiscus cannabinus L.) Mutant. Plants 2020, 9, 631. https://doi.org/10.3390/plants9050631
Lyu JI, Ramekar R, Kim D-G, Kim JM, Lee M-K, Hung NN, Kim J-B, Ahn J-W, Kang S-Y, Choi I-Y, et al. Characterization of Gene Isoforms Related to Cellulose and Lignin Biosynthesis in Kenaf (Hibiscus cannabinus L.) Mutant. Plants. 2020; 9(5):631. https://doi.org/10.3390/plants9050631
Chicago/Turabian StyleLyu, Jae Il, Rahul Ramekar, Dong-Gun Kim, Jung Min Kim, Min-Kyu Lee, Nguyen Ngoc Hung, Jin-Baek Kim, Joon-Woo Ahn, Si-Yong Kang, Ik-Young Choi, and et al. 2020. "Characterization of Gene Isoforms Related to Cellulose and Lignin Biosynthesis in Kenaf (Hibiscus cannabinus L.) Mutant" Plants 9, no. 5: 631. https://doi.org/10.3390/plants9050631
APA StyleLyu, J. I., Ramekar, R., Kim, D.-G., Kim, J. M., Lee, M.-K., Hung, N. N., Kim, J.-B., Ahn, J.-W., Kang, S.-Y., Choi, I.-Y., Park, K.-C., & Kwon, S.-J. (2020). Characterization of Gene Isoforms Related to Cellulose and Lignin Biosynthesis in Kenaf (Hibiscus cannabinus L.) Mutant. Plants, 9(5), 631. https://doi.org/10.3390/plants9050631