Co-regulation of Clustered and Neo-functionalized Genes in Plant-Specialized Metabolism

Abstract

1. Introduction
2. Neighboring Genes of Plant Specialized Metabolism
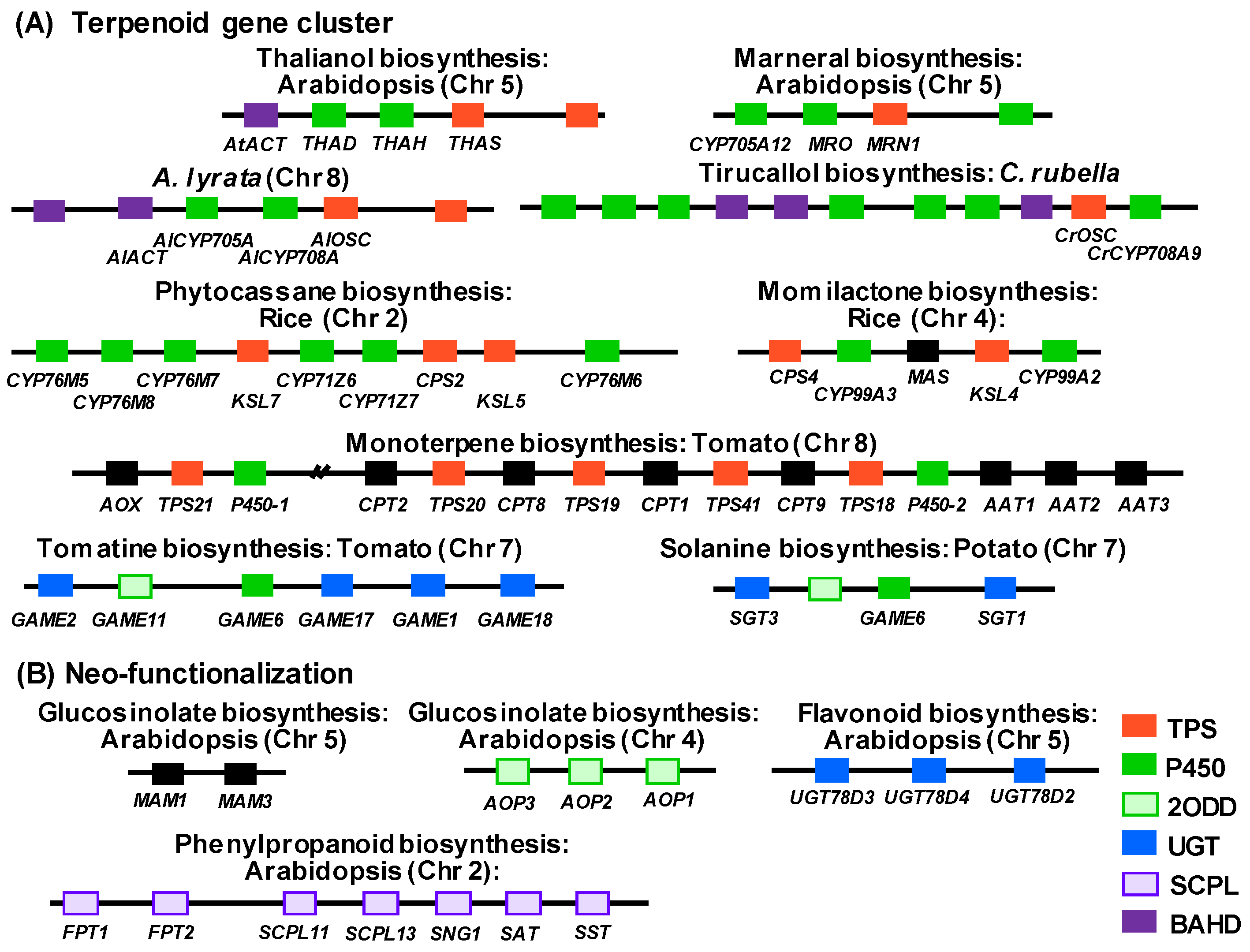
2.1. Gene Clusters Found in Plant Specialized Metabolism
2.2. Neo-functionalization Following Tandem Gene Duplication
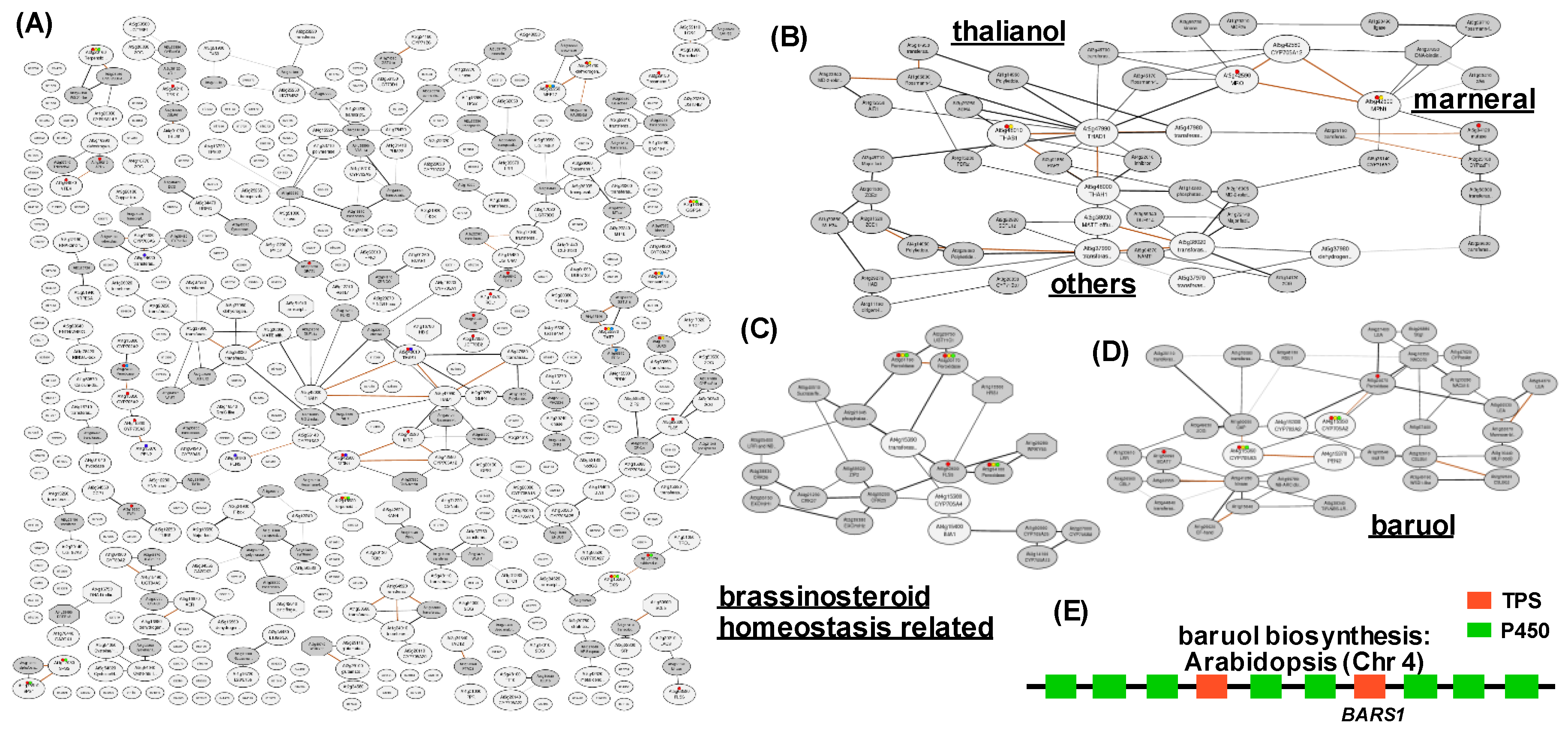
2.3. Co-expression Networks of Neighboring Genes for the Discovery of Metabolic Cluster Genes and Neo-functionalized Genes
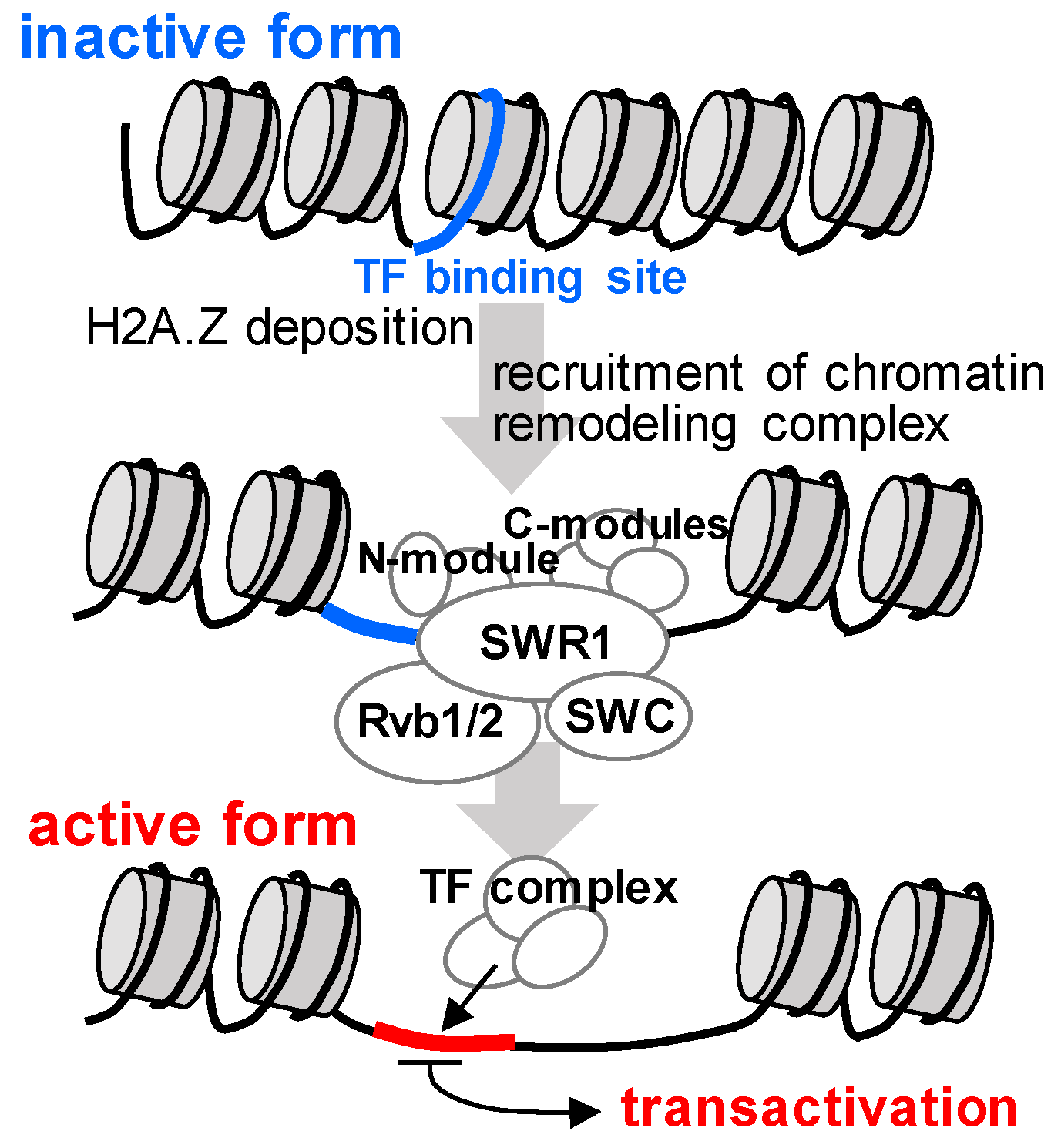
2.4. Mechanisms by Which Clustered Genes are Co-Expressed
3. Concluding Remarks and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. Shikimate and Phenylalanine Biosynthesis in the Green Lineage. Front. Plant Sci. 2013, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. The evolution of phenylpropanoid metabolism in the green lineage. Crit. Rev. Biochem. Mol. Boil. 2013, 48, 123–152. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Field, B.; Osbourn, A.E. Metabolic Diversification--Independent Assembly of Operon-Like Gene Clusters in Different Plants. Science 2008, 320, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Brotman, Y.; Lima, F.D.A.E.; Willmitzer, L.; Nikoloski, Z.; Tohge, T.; Fernie, A.R. Genomics-based strategies for the use of natural variation in the improvement of crop metabolism. Plant Sci. 2016, 242, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Nützmann, H.-W.; Scazzocchio, C.; Osbourn, A. Metabolic Gene Clusters in Eukaryotes. Annu. Rev. Genet. 2018, 52, 159–183. [Google Scholar] [CrossRef]
- Fernie, A.R.; Tohge, T. The Genetics of Plant Metabolism. Annu. Rev. Genet. 2017, 51, 287–310. [Google Scholar] [CrossRef]
- Ames, B.N.; Hartman, P.E. The Histidine Operon. Cold Spring Harb. Symp. Quant. Boil. 1963, 28, 349–356. [Google Scholar] [CrossRef]
- Shimura, K.; Okada, A.; Okada, K.; Jikumaru, Y.; Ko, K.-W.; Toyomasu, T.; Sassa, T.; Hasegawa, M.; Kodama, O.; Shibuya, N.; et al. Identification of a Biosynthetic Gene Cluster in Rice for Momilactones. J. Boil. Chem. 2007, 282, 34013–34018. [Google Scholar] [CrossRef]
- Swaminathan, S.; Morrone, D.; Wang, Q.; Fulton, D.B.; Peters, R.J. CYP76M7 is an ent-cassadiene C11alpha-hydroxylase defining a second multifunctional diterpenoid biosynthetic gene cluster in rice. Plant Cell 2009, 21, 3315–3325. [Google Scholar] [CrossRef]
- Miyamoto, K.; Fujita, M.; Shenton, M.R.; Akashi, S.; Sugawara, C.; Sakai, A.; Horie, K.; Hasegawa, M.; Kawaide, H.; Mitsuhashi, W.; et al. Evolutionary trajectory of phytoalexin biosynthetic gene clusters in rice. Plant J. 2016, 87, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Itkin, M.; Heinig, U.; Tzfadia, O.; Bhide, A.J.; Shinde, B.; Cárdenas, P.D.; Bocobza, S.E.; Unger, T.; Malitsky, S.; Finkers, R.; et al. Biosynthesis of Antinutritional Alkaloids in Solanaceous Crops Is Mediated by Clustered Genes. Science 2013, 341, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Matsuba, Y.; Nguyen, T.T.; Wiegert, K.; Falara, V.; Gonzales-Vigil, E.; Leong, B.; Schäfer, P.; Kudrna, D.; A Wing, R.; Bolger, A.M.; et al. Evolution of a Complex Locus for Terpene Biosynthesis in Solanum[W][OPEN]. Plant Cell 2013, 25, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Bakht, S.; Leggett, M.; Maxwell, C.; Melton, R.; Osbourn, A. A gene cluster for secondary metabolism in oat: Implications for the evolution of metabolic diversity in plants. Proc. Natl. Acad. Sci. USA 2004, 101, 8233–8238. [Google Scholar] [CrossRef]
- Field, B.; Fiston-Lavier, A.-S.; Kemen, A.; Geisler, K.; Quesneville, H.; Osbourn, A.E. Formation of plant metabolic gene clusters within dynamic chromosomal regions. Proc. Natl. Acad. Sci. USA 2011, 108, 16116–161121. [Google Scholar] [CrossRef]
- Nützmann, H.-W.; Osbourn, A. Regulation of metabolic gene clusters in Arabidopsis thaliana. New Phytol. 2014, 205, 503–510. [Google Scholar] [CrossRef]
- Liu, Z.; Duran, H.G.S.; Harnvanichvech, Y.; Stephenson, M.; Schranz, M.E.; Nelson, D.; Medema, M.H.; Osbourn, A. Drivers of metabolic diversification: How dynamic genomic neighbourhoods generate new biosynthetic pathways in the Brassicaceae. New Phytol. 2019. [Google Scholar] [CrossRef]
- Krokida, A.; Delis, C.; Geisler, K.; Garagounis, C.; Tsikou, D.; Peña-Rodríguez, L.M.; Katsarou, D.; Field, B.; Osbourn, A.E.; Papadopoulou, K.K. A metabolic gene cluster inLotus japonicusdiscloses novel enzyme functions and products in triterpene biosynthesis. New Phytol. 2013, 200, 675–690. [Google Scholar] [CrossRef]
- Boycheva, S.; Daviet, L.; Wolfender, J.-L.; Fitzpatrick, T.B. The rise of operon-like gene clusters in plants. Trends Plant Sci. 2014, 19, 447–459. [Google Scholar] [CrossRef]
- Kliebenstein, D.J. Genetic Control of Natural Variation in Arabidopsis Glucosinolate Accumulation. Plant Physiol. 2001, 126, 811–825. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Lambrix, V.M.; Reichelt, M.; Gershenzon, J.; Mitchell-Olds, T. Gene Duplication in the Diversification of Secondary Metabolism: Tandem 2-Oxoglutarate–Dependent Dioxygenases Control Glucosinolate Biosynthesis in Arabidopsis. Plant Cell 2001, 13, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.; Hansen, L.G.; Mirza, N.; Crocoll, C.; Mirza, O.A.; Halkier, B.A. Changing substrate specificity and iteration of amino acid chain elongation in glucosinolate biosynthesis through targeted mutagenesis of Arabidopsis methylthioalkylmalate synthase. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Wendenburg, R.; Ishihara, H.; Nakabayashi, R.; Watanabe, M.; Suplice, R.; Hoefgen, R.; Takayama, H.; Saito, K.; Stitt, M.; et al. Characterization of a recently evolved flavonol-phenylacyltransferase gene provides signatures of natural light selection in Brassicaceae. Nat. Commun. 2016, 7, 12399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Alseekh, S.; Fernie, A.R. Metabolomics 20 years on: What have we learned and what hurdles remain? Plant J. 2018, 94, 933–942. [Google Scholar] [CrossRef]
- Wang, S.; Alseekh, S.; Fernie, A.R.; Luo, J. The Structure and Function of Major Plant Metabolite Modifications. Mol. Plant 2019, 12, 899–919. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Töpfer, N.; Fuchs, L.-M.; Aharoni, A. The PhytoClust tool for metabolic gene clusters discovery in plant genomes. Nucleic Acids Res. 2017, 45, 7049–7063. [Google Scholar] [CrossRef]
- A Kautsar, S.; Duran, H.G.S.; Blin, K.; Osbourn, A.; Medema, M.H. plantiSMASH: Automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, W55–W63. [Google Scholar] [CrossRef]
- Williams, E.J.; Bowles, D.J. Coexpression of Neighboring Genes in the Genome of Arabidopsis thaliana. Genome Res. 2004, 14, 1060–1067. [Google Scholar] [CrossRef]
- Medema, M.H.; Osbourn, A. Computational genomic identification and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep. 2016, 33, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.-Z.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the Fruit Metabolome in Tomato Breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; De Souza, L.P.; Fernie, A.R. On the natural diversity of phenylacylated-flavonoid and their in planta function under conditions of stress. Phytochem. Rev. 2017, 17, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, T.P.; Schnitzler, J.-P.; Heller, W.; Sandermann, H.; Hertkorn, N.; Szymczak, W.; Metzger, J.W. Structures of UV-B Induced Sunscreen Pigments of the Scots Pine (Pinus sylvestris L.). Angew. Chem. Int. Ed. 1995, 34, 312–314. [Google Scholar] [CrossRef]
- Peng, M.; Shahzad, R.; Gul, A.; Subthain, H.; Shen, S.; Lei, L.; Zheng, Z.; Zhou, J.; Lu, D.; Wang, S.; et al. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 2017, 8, 1975. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yuan, H.; Dong, X.; Peng, M.; Jing, X.; Xu, Q.; Tang, T.; Wang, Y.; Zha, S.; Gao, M.; et al. Genome-wide Dissection of Co-selected UV-B Responsive Pathways in the UV-B Adaptation of Qingke. Mol. Plant 2019, 13, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; De Souza, L.P.; Benina, M.; Fernie, A.R. The style and substance of plant flavonoid decoration; towards defining both structure and function. Phytochemistry 2020, 174, 112347. [Google Scholar] [CrossRef]
- Luo, J.; Nishiyama, Y.; Fuell, C.; Taguchi, G.; Elliott, K.; Hill, L.; Tanaka, Y.; Kitayama, M.; Yamazaki, M.; Bailey, P.; et al. Convergent evolution in the BAHD family of acyl transferases: Identification and characterization of anthocyanin acyl transferases from Arabidopsis thaliana. Plant J. 2007, 50, 678–695. [Google Scholar] [CrossRef]
- Fraser, C.M.; Thompson, M.G.; Shirley, A.M.; Ralph, J.; Schoenherr, J.A.; Sinlapadech, T.; Hall, M.C.; Chapple, C. Related Arabidopsis Serine Carboxypeptidase-Like Sinapoylglucose Acyltransferases Display Distinct But Overlapping Substrate Specificities1[OA]. Plant Physiol. 2007, 144, 1986–1999. [Google Scholar] [CrossRef]
- Stehle, F.; Brandt, W.; Stubbs, M.; Milkowski, C.; Strack, D. Sinapoyltransferases in the light of molecular evolution. Phytochemistry 2009, 70, 1652–1662. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, B.; Qi, Y.-W.; Liu, C.; Liu, Z.; Ren, X. Biochemical and functional characterization of AcUFGT3a, a galactosyltransferase involved in anthocyanin biosynthesis in the red-fleshed kiwifruit (Actinidia chinensis). Physiol. Plant. 2017, 162, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Zhang, Y.; Peterek, S.; Matros, A.; Rallapalli, G.; Tandrón, Y.A.; Butelli, E.; Kallam, K.; Hertkorn, N.; Mock, H.; et al. Ectopic expression of snapdragon transcription factors facilitates the identification of genes encoding enzymes of anthocyanin decoration in tomato. Plant J. 2015, 83, 686–704. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Fernie, A.R. Annotation of Plant Gene Function via Combined Genomics, Metabolomics and Informatics. J. Vis. Exp. 2012, e3487. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Fernie, A.R. Co-expression and co-responses: Within and beyond transcription. Front. Plant Sci. 2012, 3, 3. [Google Scholar] [CrossRef]
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.M.; Wilkins, O.; Campbell, M.M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined Sequence and Expression Comparisons across Plant Networks Derived from Seven Species[W][OA]. Plant Cell 2011, 23, 895–910. [Google Scholar] [CrossRef]
- Mutwil, M.; Øbro, J.; Willats, W.; Persson, S. GeneCAT--novel webtools that combine BLAST and co-expression analyses. Nucleic Acids Res. 2008, 36, W320–W326. [Google Scholar] [CrossRef]
- Tohge, T.; Fernie, A.R. Lignin, mitochondrial family, and photorespiratory transporter classification as case studies in using co-expression, co-response, and protein locations to aid in identifying transport functions. Front. Plant Sci. 2014, 5, 5. [Google Scholar] [CrossRef]
- Araujo, W.; Tohge, T.; Ishizaki, K.; Leaver, C.J.; Fernie, A.R. Protein degradation – an alternative respiratory substrate for stressed plants. Trends Plant Sci. 2011, 16, 489–498. [Google Scholar] [CrossRef]
- Wada, M.; Takahashi, H.; Amin, A.-U.; Nakamura, K.; Hirai, M.Y.; Ohta, D.; Kanaya, S. Prediction of operon-like gene clusters in the Arabidopsis thaliana genome based on co-expression analysis of neighboring genes. Gene 2012, 503, 56–64. [Google Scholar] [CrossRef]
- Takos, A.; Knudsen, C.; Lai, D.; Kannangara, R.; Mikkelsen, L.; Motawia, M.S.; Olsen, C.E.; Sato, S.; Tabata, S.; Jørgensen, K.; et al. Genomic clustering of cyanogenic glucoside biosynthetic genes aids their identification in Lotus japonicus and suggests the repeated evolution of this chemical defence pathway. Plant J. 2011, 68, 273–286. [Google Scholar] [CrossRef]
- Obayashi, T.; Kinoshita, K.; Nakai, K.; Shibaoka, M.; Hayashi, S.; Saeki, M.; Shibata, D.; Saito, K.; Ohta, H. ATTED-II: A database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 2006, 35, D863–D869. [Google Scholar] [CrossRef]
- Obayashi, T.; Hayashi, S.; Saeki, M.; Ohta, H.; Kinoshita, K. ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res. 2008, 37, D987–D991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, L. Arabidopsis BRASSINOSTEROID INACTIVATOR2 is a typical BAHD acyltransferase involved in brassinosteroid homeostasis. J. Exp. Bot. 2018, 69, 1925–1941. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Karkute, S.; Banerjee, S.; Meena, N.L.; Dahuja, A. Contemporary Understanding of miRNA-Based Regulation of Secondary Metabolites Biosynthesis in Plants. Front. Plant Sci. 2017, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Li, Y.; Li, C.; Zhang, Y. Differential microRNA Analysis of Glandular Trichomes and Young Leaves in Xanthium strumarium L. Reveals Their Putative Roles in Regulating Terpenoid Biosynthesis. PLOS ONE 2015, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Shan, J.-X.; Shi, M.; Gao, J.-P.; Lin, H.-X. ThemiR156-SPL9-DFRpathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef]
- Rokas, A.; Wisecaver, J.H.; Lind, A. The birth, evolution and death of metabolic gene clusters in fungi. Nat. Rev. Genet. 2018, 16, 731–744. [Google Scholar] [CrossRef]
- Fernie, A.R.; Tohge, T. Location, location, location--no more! The unravelling of chromatin remodeling regulatory aspects of plant metabolic gene clusters. New Phytol. 2015, 205, 458–460. [Google Scholar] [CrossRef]
- Nützmann, H.-W.; Osbourn, A. Gene clustering in plant specialized metabolism. Curr. Opin. Biotechnol. 2014, 26, 91–99. [Google Scholar] [CrossRef]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved Histone Variant H2A.Z Protects Euchromatin from the Ectopic Spread of Silent Heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef]
- Sarnowska, E.A.; Rolicka, A.; Bucior, E.; Cwiek, P.; Tohge, T.; Fernie, A.R.; Jikumaru, Y.; Kamiya, Y.; Franzen, R.; Schmelzer, E.; et al. DELLA-Interacting SWI3C Core Subunit of Switch/Sucrose Nonfermenting Chromatin Remodeling Complex Modulates Gibberellin Responses and Hormonal Cross Talk in Arabidopsis1[W]. Plant Physiol. 2013, 163, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Sarnowska, E.; Gratkowska, D.M.; Sacharowski, S.P.; Cwiek, P.; Tohge, T.; Fernie, A.R.; Siedlecki, J.A.; Koncz, C.; Sarnowski, T.J. The Role of SWI/SNF Chromatin Remodeling Complexes in Hormone Crosstalk. Trends Plant Sci. 2016, 21, 594–608. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Species | Annotated Metabolic Gene Cluster | Tandem Gene Duplication of Single Gene Family | ||||
|---|---|---|---|---|---|---|
| P450 | 2ODD | TPS | PKS | UGT | ||
| A. thaliana | 39 | 28 | 16 | 6 | 5 | 15 |
| O. sativa | 34 | 57 | 15 | 10 | 10 | 37 |
| S. lycopersicum | 50 | 30 | 24 | 8 | 9 | 30 |
| L. japonicus | 18 | 29 | 7 | 5 | 4 | 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tohge, T.; Fernie, A.R. Co-regulation of Clustered and Neo-functionalized Genes in Plant-Specialized Metabolism. Plants 2020, 9, 622. https://doi.org/10.3390/plants9050622
Tohge T, Fernie AR. Co-regulation of Clustered and Neo-functionalized Genes in Plant-Specialized Metabolism. Plants. 2020; 9(5):622. https://doi.org/10.3390/plants9050622
Chicago/Turabian StyleTohge, Takayuki, and Alisdair R. Fernie. 2020. "Co-regulation of Clustered and Neo-functionalized Genes in Plant-Specialized Metabolism" Plants 9, no. 5: 622. https://doi.org/10.3390/plants9050622
APA StyleTohge, T., & Fernie, A. R. (2020). Co-regulation of Clustered and Neo-functionalized Genes in Plant-Specialized Metabolism. Plants, 9(5), 622. https://doi.org/10.3390/plants9050622