Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship


Abstract
1. Introduction
2. Results and Discussion
2.1. Genome Features of B. chinense and B. commelynoideum
2.2. Codon Usage Bias Analysis
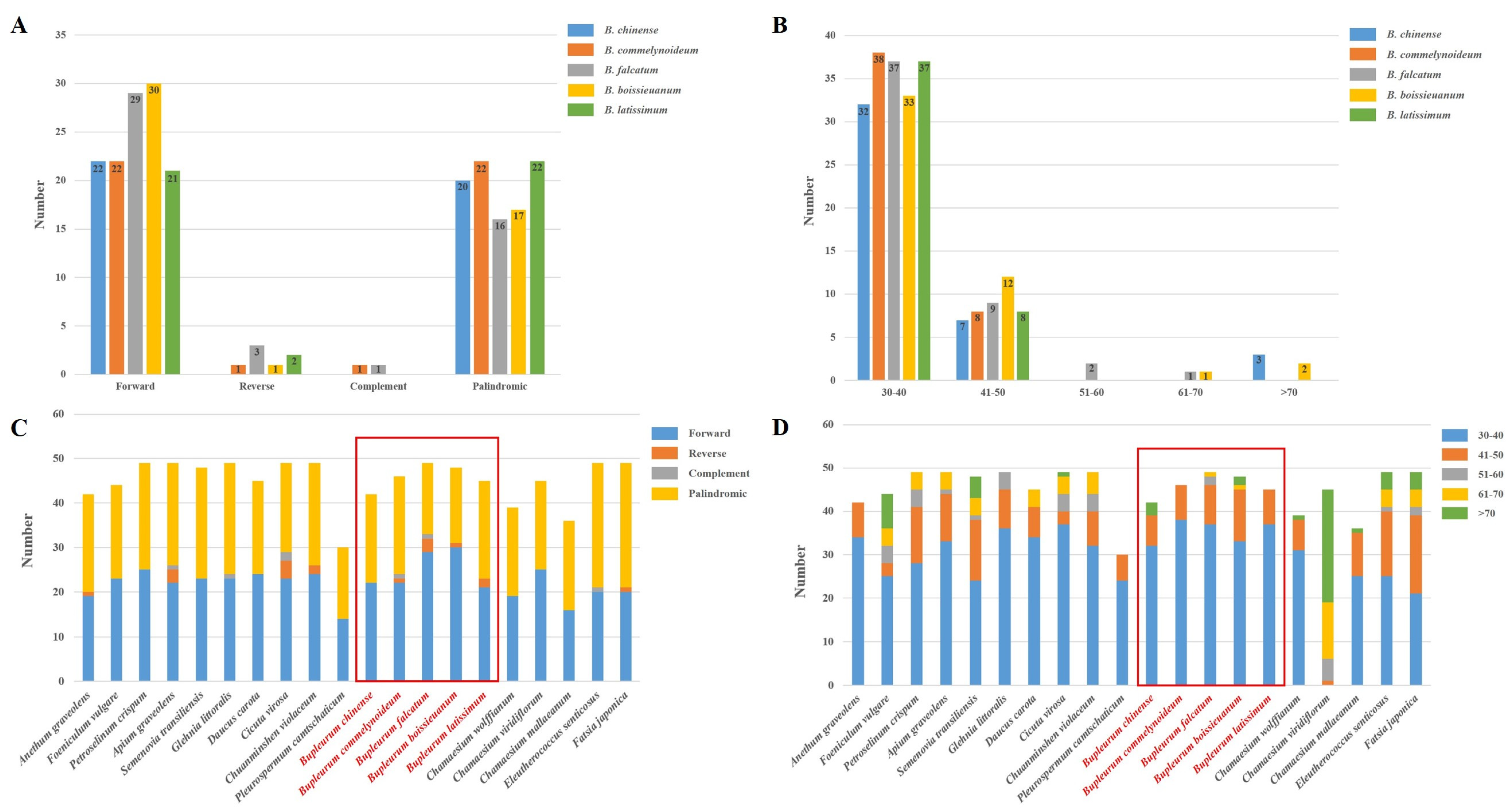
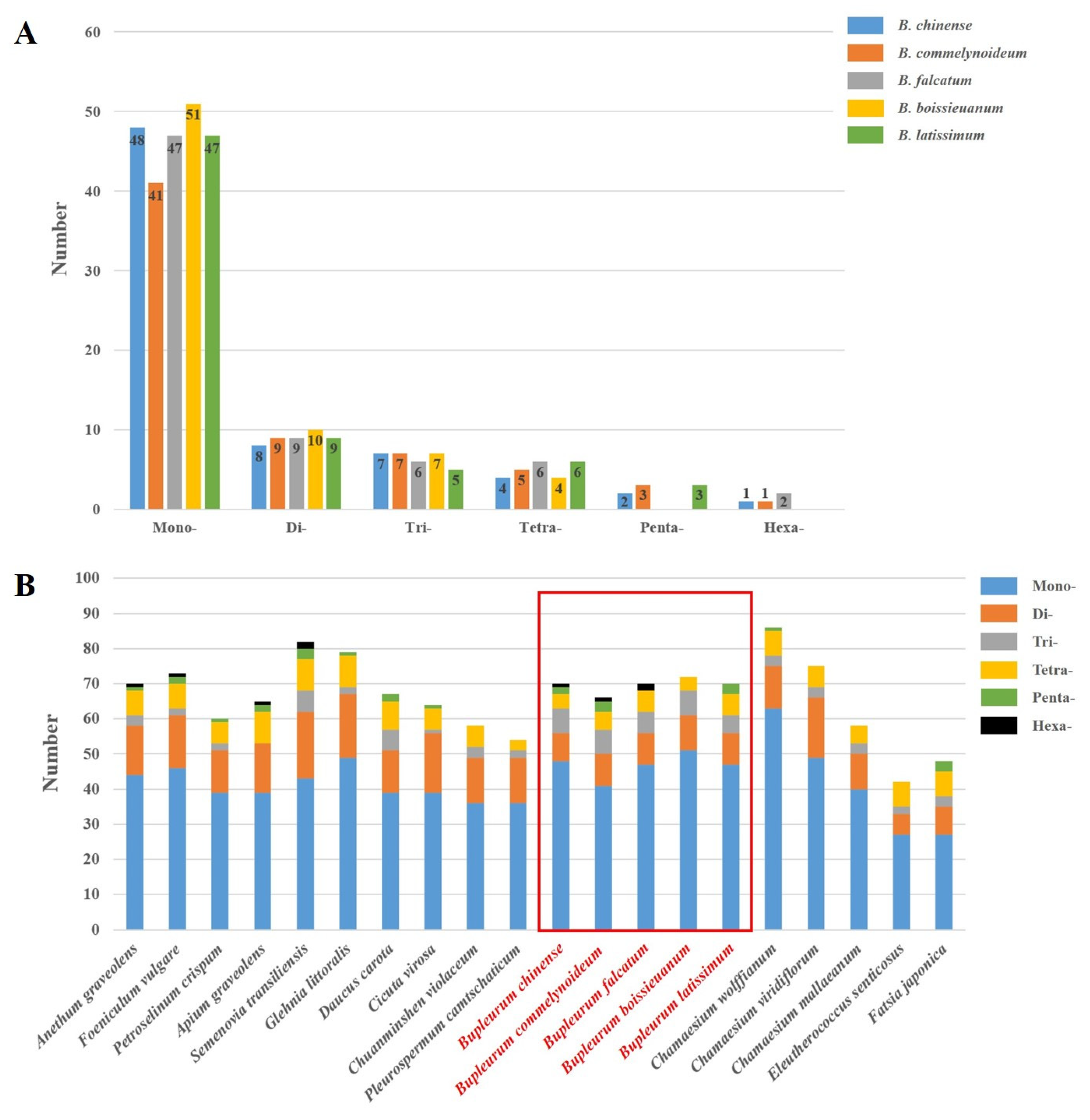
2.3. Repetitive Sequences Analysis
2.4. Nucleotide Diversity Analysis
2.5. Selective Pressure Analysis
2.6. IR Boundary Comparative Analysis
2.7. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Materials and DNA Extraction
3.2. Genome Sequencing, Assembly and Annotation
3.3. Codon Usage Bias Analysis
3.4. Repetitive Sequences Analysis
3.5. Nucleotide Diversity Analysis
3.6. Selective Pressure Analysis
3.7. IR Boundary Comparative Analysis
3.8. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- She, M.L.; Watson, M.F. Apiaceae (Umbelliferae). In Flora of China; Science Press: Beijing, China, 2005; Volume 14, pp. 60–74. [Google Scholar]
- Pan, S.L. Bupleurum Species: Scientific Evaluation and Clinical Applications. In Traditional Herbal Medicines for Modern Times; CRC Press: Boca Raton, FL, USA, 2006; Volume 107, pp. 13–16. [Google Scholar]
- Chinese Pharmacopoeia Commission. Bupleuri Radix. In Pharmacopoeia of People’s Republic of China; China Medical Science Press: Beijing, China, 2015; Volume 5, pp. 280–281. [Google Scholar]
- Guo, M.N.; Liu, S.X.; Zhao, Y.M.; Zhang, T.J.; Liu, D.L. Analysis on chemical constituents in Bupleuri Radix by HPLC-Q-TOF-MS. Chin. Tradit. Herb. Drugs 2016, 47, 2044–2052. [Google Scholar]
- Huang, H.Q.; Wang, X.H.; Fu, H.; Wang, Y.; Yang, S.H. Research progress on medicinal plant resources of Bupleurum L. Chin. Tradit. Herb. Drugs 2017, 48, 2989–2996. [Google Scholar]
- Cholet, J.; Decombat, C.; Vareille-Delarbre, M.; Gainche, M.; Berry, A.; Senejoux, F.; Ripoche, I.; Delort, L.; Vermerie, M.; Fraisse, D.; et al. In vitro anti-inflammatory and immunomodulatory activities of an extract from the roots of Bupleurum rotundifolium. Medicines 2019, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.P.; Lan, K.L.; Liao, S.X.; Huang, Y.H.; Hou, M.C.; Lan, K.H. Antiviral effect of saikosaponin B2 in combination with daclatasvir on NS5A resistance-associated substitutions of hepatitis C virus. J. Chin. Med. Assoc. 2019, 82, 368–374. [Google Scholar] [CrossRef]
- Zhao, J.F.; Guo, Y.Z.; Meng, X.S. The toxic principles of Bupleurum longiradiatum. Acta Pharm. Sinica 1987, 22, 507–511. [Google Scholar]
- Liu, X.J.; Hu, J.; Li, Z.Y.; Qin, X.M.; Zhang, L.Z.; Guo, X.Q. Species classification and quality assessment of Chaihu (Radix Bupleuri) based on high-performance liquid chromatographic fingerprint and combined chemometrics methods. Arch. Pharm. Res. 2011, 34, 961–969. [Google Scholar] [CrossRef]
- Downie, S.R.; Spalik, K.; Katz-Downie, D.S.; Reduron, J. Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant. Div. Evol. 2010, 128, 111–136. [Google Scholar] [CrossRef]
- Zhou, J.; Gong, X.; Downie, S.R.; Peng, H. Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: Additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Mol. Phylogenet. Evol. 2009, 53, 56–68. [Google Scholar] [CrossRef]
- Neves, S.S.; Watson, M.F. Phylogenetic relationships in Bupleurum (Apiaceae) based on nuclear ribosomal DNA ITS sequence data. Ann. Bot. 2004, 93, 379–398. [Google Scholar] [CrossRef]
- Wang, Q.Z.; He, X.J.; Zhou, S.D.; Wu, Y.K.; Yu, Y.; Pang, Y.L. Phylogenetic inference of the genus Bupleurum (Apiaceae) in Hengduan Mountains based on chromosome counts and nuclear ribosomal DNA ITS sequences. J. Syst. Evol. 2008, 46, 142–154. [Google Scholar]
- Wang, C.B.; Ma, X.G.; He, X.J. A taxonomic re-ssessment in the Chinese Bupleurum (Apiaceae): Insights from morphology, nuclear ribosomal internal transcribed spacer, and chloroplast (trnH-psbA, matK) sequences. J. Syst. Evol. 2011, 49, 558–589. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lee, J.H.; Kang, S.H.; Ahn, B.O.; Kim, C.K. The complete chloroplast genome of the hare’s ear root, Bupleurum falcatum: Its molecular features. Genes 2016, 7, 20. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, T.Z.; Qiu, D.Y.; Chai, Q.; Fan, W.B.; Li, Z.H.; Fang, M.F. Complete plastid genome of Bupleurum boissieuanum, an endemic herb plant in western China. Conserv. Genet. Resour. 2018, 10, 635–637. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast genome. Plant Mol. Biol. 1992, 19, 149–168. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, J.; Jia, Y.; Li, W.L.; Xu, F.S.; Wang, X.M. Comparative chloroplast genome analyses of species in Gentiana section Cruciata (Gentianaceae) and the development of authentication markers. Int. J. Mol. Sci. 2018, 19, 1962. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, D.Z. Advances in phylogenomics based on complete chloroplast genomes. Plant Divers. Resour. 2011, 33, 365–375. [Google Scholar]
- Krak, K.; Vít, P.; Belyayev, A.; Douda, J.; Hreusová, L.; Mandák, B. Allopolyploid origin of Chenopodium album s. str. (Chenopodiaceae): A molecular and cytogenetic insight. PLoS ONE 2016, 11, e0161063. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Y.; Liu, Y.M.; Xie, D.F.; He, X.J.; Zhou, S.D. Comparative chloroplast genomics of Fritillaria (Liliaceae), inferences for phylogenetic relationships between Fritillaria and Lilium and plastome evolution. Plants 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.Y.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; dePamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the Felsenstein Zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef]
- Xie, D.F.; Yu, Y.; Deng, Y.Q.; Li, J.; Liu, H.Y.; Zhou, S.D.; He, X.J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Zhu, A.D.; Guo, W.H.; Gupta, S.; Fan, W.S.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.Y.; Gao, H.H.; Zhu, Y.J.; Xu, J.; Pang, X.H.; Yao, H.; Sun, C.; Li, X.E.; Li, C.Y.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.W.; Lee, I.J. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef]
- Gu, C.H.; Tembrock, L.R.; Zheng, S.Y.; Wu, Z.Q. The complete chloroplast genome of Catha edulis: A comparative analysis of genome features with related species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.W.; Liu, Q.Q.; Chen, W.N.; Li, F.; Chen, A.M.; Li, C.; Chao, Z. Characterization of complete chloroplast genome of Angelica sinensis (Apiaceae), an endemic medical plant to China. Mitochondrial DNA B Resour. 2019, 4, 158–159. [Google Scholar] [CrossRef]
- Zhu, Q.L.; Guang, Y.L.; Xiao, X.F.; Shan, N.; Wan, C.P.; Yang, Y.X. The complete chloroplast genome sequence of the Apium graveolens L. (Apiaceae). Mitochondrial DNA B Resour. 2019, 4, 463–464. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Guo, X.L.; He, X.J.; Yu, Y.; Zhou, S.D. The complete chloroplast genome of Chamaesium paradoxum. Mitochondrial DNA B Resour. 2019, 4, 2069–2070. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Cho, S.T.; Haryono, M.; Kuo, C.H. Complete chloroplast genome sequence of common bermudagrass (Cynodon dactylon (L.) Pers.) and comparative analysis within the family Poaceae. PLoS ONE 2017, 12, e0179055. [Google Scholar]
- Logacheva, M.D.; Shipunov, A.B. Phylogenomic analysis of Picramnia, Alvaradoa, and Leitneria supports the independent Picramniales. J. Syst. Evol. 2017, 55, 171–176. [Google Scholar] [CrossRef]
- Vanin, E.F. Processed pseudogenes: Characteristics and evolution. Ann. Rev. Genet. 1985, 19, 253–272. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef]
- Morton, B.R. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Kim, K.J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.H.; Shang, A.Q.; Zhang, S.; Yu, X.Y.; Ren, Y.C.; Yang, M.S.; Wang, J.M. The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: Genome comparative and taxonomic position analysis. PLoS ONE 2017, 12, e0171264. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Bio. 2017, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. 2012, 88, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; de Pouplana, L.R. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet. 2012, 28, 574–581. [Google Scholar] [CrossRef]
- Ren, G.P.; Dong, Y.Y.; Dang, Y.K. Codon codes: Codon usage bias influences many levels of gene expression. Sci. Chine Life Sci. 2019, 49, 839–847. [Google Scholar]
- Khan, A.; Asaf, S.; Khan, A.L.; Shehzad, T.; Al-Rawahi, A.; Al-Harrasi, A. Comparative chloroplast genomics of endangered Euphorbia species: Insights into hotspot divergence, repetitive sequence variation, and phylogeny. Plants 2020, 9, 199. [Google Scholar] [CrossRef]
- Huang, J.; Chen, R.H.; Li, X.G. Comparative analysis of the complete chloroplast genome of four known Ziziphus species. Genes 2017, 8, 340. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Chloroplast evolution: Secondary dispatch symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yu, Y.; Deng, Y.Q.; Li, J.; Huang, Z.X.; Zhou, S.D. The chloroplast genome of Lilium henrici: Genome structure and comparative analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Yang, Y.C.; Zhou, T.; Liu, Z.L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G.F. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Wang, W.B.; Yu, H.; Wang, J.H.; Lei, W.J.; Gao, J.H.; Qiu, X.P.; Wang, J.S. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.M.; Khan, M.A.; Lee, S.M.; Lee, I.J. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 2016, 7, 447. [Google Scholar] [CrossRef]
- Rose, O.; Falush, D. A threshold size for microsatellite expansion. Mol. Biol. Evol. 1998, 15, 613–615. [Google Scholar] [CrossRef]
- Kaur, S.; Panesar, P.S.; Bera, M.B.; Kaur, V. Simple sequence repeat markers in genetic divergence and marker-assisted selection of rice cultivars: A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 41–49. [Google Scholar] [CrossRef]
- Chung, S.M.; Staub, J.E.; Chen, J.F. Molecular phylogeny of Cucumis species as revealed by consensus chloroplast SSR marker length and sequence variation. Genome 2006, 49, 219–229. [Google Scholar] [CrossRef]
- Chen, S.L.; Song, J.Y.; Yao, H.; Shi, L.C.; Luo, K.; Han, J.P. Strategy and key technique of identification of Chinese herbal medicine using DNA barcoding. Chin. J. Nat. Med. 2009, 7, 322–327. [Google Scholar] [CrossRef]
- Song, C.F.; Wu, B.C.; Zhou, W.; Liu, Q.X. Analyses on relationship and taxonomic position of Chuanminshen Sheh et Shan (Apiaceae) based on variation of psbA-trnH sequence. J. Plant Resour. Environ. 2014, 23, 19–26. [Google Scholar]
- Neubig, K.M.; Whitten, W.M.; Carlsward, B.S.; Blanco, M.A.; Endara, L.; Williams, N.H.; Moore, M. Phylogenetic utility of ycf1 in orchids: A plastid gene more variable than matK. Plant. Syst. Evol. 2008, 277, 75–84. [Google Scholar] [CrossRef]
- Saslis-Lagoudakis, C.H.; Klitgaard, B.B.; Forest, F.; Francis, L.; Savolainen, V.; Williamson, E.M.; Hawkins, J.A. The use of phylogeny to interpret cross-cultural patterns in plant use and guide medicinal plant discovery: An example from Pterocarpus (Leguminosae). PLoS ONE 2011, 6, e22275. [Google Scholar] [CrossRef]
- Ye, M.R.; Liu, W.; Xue, Q.Y.; Hou, B.E.; Luo, J.; Ding, X.Y. Phylogeography of the endangered orchid Dendrobium moniliforme in East Asia inferred from chloroplast DNA sequences. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2017, 28, 880–891. [Google Scholar] [PubMed]
- Yang, Z.H.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Yang, Z.H.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Gao, N.N.; Zhao, Z.L.; Ni, L.H. Prospect: Identification of medicinal plant based on chloroplast gene ycf15. Chin. Tradit. Herb. Drugs. 2017, 48, 3210–3217. [Google Scholar]
- Kang, L.; Xie, D.F.; Xiao, Q.Y.; Peng, C.; Yu, Y.; He, X.J. Sequencing and analyses on chloroplast genomes of Tetrataenium candicans and two allies give new insights on structural variants, DNA barcoding and phylogeny in Apiaceae subfamily Apioideae. PeerJ 2019, 7, e8063. [Google Scholar] [CrossRef]
- Mustafina, F.U.; Yi, D.K.; Choi, K.; Shin, C.H.; Tojibaev, K.S.; Downie, S.R. A comparative analysis of complete plastid genomes from Prangos fedtschenkoi and Prangos lipskyi (Apiaceae). Ecol. Evol. 2019, 9, 364–377. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Wang, Y.H.; Qu, X.J.; Chen, S.Y.; Li, D.Z.; Yi, T.S. Plastomes of Mimosoideae: Structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet. Genomes 2017, 13, 41. [Google Scholar] [CrossRef]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.J.; Yan, C.X.; Zhao, X.B.; Shan, S.H. A comparative analysis of the complete chloroplast genome sequences of four peanut botanical varieties. PeerJ 2018, 6, e5349. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Wang, C.B.; Wen, J.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Chamaesium (Apiaceae: Apioideae) inferred from ITS, cpDNA and morphological characters. Phytotaxa 2018, 376, 001–016. [Google Scholar] [CrossRef]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.F.; Li, Z.H. Comparative analysis of the complete chloroplast genome of four endangered herbals of Notopterygium. Genes 2017, 8, 124. [Google Scholar] [CrossRef]
- Zhang, L.; Li, M.; Zhao, J.C. Fruit morphology of 23 species in Umbelliferae and its taxonomic significance. Acta Bot. Boreal. Occident. Sin. 2015, 35, 2428–2438. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. CodonW. Ph.D. Thesis, University of Nottingham, Nottinghamshire, UK, 1999. [Google Scholar]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | B. chinense | B. commelynoideum | B. falcatum | B. boissieuanum | B. latissimum |
|---|---|---|---|---|---|
| Genome length (bp) | 155,869 | 155,629 | 155,989 | 156,108 | 155,621 |
| LSC length (bp) | 85,772 | 85,542 | 85,912 | 86,007 | 85,471 |
| SSC length (bp) | 17,501 | 17,495 | 17,517 | 17,495 | 17,548 |
| IR length (bp) | 26,298 | 26,296 | 26,280 | 26,303 | 26,300 |
| CDS length (bp) | 78,249 | 78,255 | 78,261 | 78,270 | 78,264 |
| Overall GC content (%) | 37.7 | 37.7 | 37.7 | 37.7 | 37.6 |
| LSC GC content (%) | 35.8 | 35.8 | 35.8 | 35.8 | 35.8 |
| SSC GC content (%) | 31.4 | 31.4 | 31.4 | 31.4 | 31.3 |
| IR GC content (%) | 42.8 | 42.8 | 42.8 | 42.8 | 42.8 |
| CDS GC content (%) | 38.3 | 38.2 | 38.3 | 38.3 | 38.2 |
| Groups | Categories | Name of Genes | Number |
|---|---|---|---|
| Self-replication | rRNAs | rrn4.5(×2); rrn5(×2); rrn16(×2); rrn23(×2) | 8 |
| tRNAs | trnA-UGC(×2); trnC-GCA; trnD-GUC; trnE-UUC; trnF-GAA; trnfM-CAU; trnG-GCC; trnG-UCC; trnH-GUG; trnI-CAU(×2); trnI-GAU(×2); trnK-UUU; trnL-CAA(×2); trnL-UAA; trnL-UAG; trnM-CAU; trnN-GUU(×2); trnP-UGG; trnQ-UUG; trnR-ACG(×2); trnR-UCU; trnS-GCU; trnS-GGA; trnS-UGA; trnT-GGU; trnT-UGU; trnV-GAC(×2); trnV-UAC; trnW-CCA; trnY-GUA | 37 | |
| DNA-dependent RNA polymerase | rpoA; rpoB; rpoC1; rpoC2 | 4 | |
| Small subunit of ribosomal proteins | rps2; rps3; rps4; rps7(×2); rps8; rps11; rps12(×2); rps14; rps15; rps16; rps18; rps19 | 14 | |
| Large subunit of ribosomal proteins | rpl2(×2); rpl14; rpl16; rpl20; rpl22; rpl23(×2); rpl3; rpl33; rpl36 | 11 | |
| Genes for photosynthesis | Subunits of NADH dehydrogenase | ndhA; ndhB(×2); ndhC; ndhD; ndhE; ndhF; ndhG; ndhH; ndhI; ndhJ; ndhK | 12 |
| Subunits of photosystem Ⅰ | psaA; psaB; psaC; psaI; psaJ; ycf3; ycf4 | 7 | |
| Subunits of photosystem Ⅱ | psbA; psbB; psbC; psbD; psbE; psbF; psbH; psbI; psbJ; psbK; psbL; psbM; psbN; psbT; psbZ | 15 | |
| Subunits of cytochrome b/f complex | petA; petB; petD; petG; petL; petN | 6 | |
| Subunits of ATP synthase | atpA; atpB; atpE; atpF; atpH; atpI | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Other genes | ATP-dependent protease subunit P | clpP | 1 |
| Maturase | matK | 1 | |
| Subunits of Acetyl-CoA-carboxylase | accD | 1 | |
| Envelop membrane protein | cemA | 1 | |
| C-type cytochrome synthesis gene | ccsA | 1 | |
| Genes of unknown function | Hypothetical chloroplast reading frames | ycf1; ycf2(×2) | 3 |
| Pseudogenes | infA; ycf15; ycf1*; rps19 * | 4 | |
| Total | 114 single-copy genes, 133 in total. | ||
| Index | B. chinense | B. commelynoideum | B. falcatum | B. boissieuanum | B. latissimum |
|---|---|---|---|---|---|
| Length (bp) | 63,564 | 63,570 | 63,576 | 63,585 | 63,579 |
| Codon No. | 21,188 | 21,190 | 21,192 | 21,195 | 21,193 |
| Amino acid No. | 21,135 | 21,137 | 21,139 | 21,142 | 21,140 |
| SC No. | 20,273 | 20,274 | 20,275 | 20,279 | 20,276 |
| ENC | 49.90 | 49.87 | 49.86 | 49.88 | 49.83 |
| CAI | 0.166 | 0.166 | 0.167 | 0.167 | 0.166 |
| CBI | −0.100 | −0.101 | −0.100 | −0.100 | −0.102 |
| FOP | 0.354 | 0.354 | 0.354 | 0.354 | 0.353 |
| GC content (%) | 0.382 | 0.382 | 0.382 | 0.382 | 0.382 |
| GC3 content (%) | 0.269 | 0.269 | 0.269 | 0.269 | 0.269 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xie, D.-F.; Guo, X.-L.; Zheng, Z.-Y.; He, X.-J.; Zhou, S.-D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants 2020, 9, 543. https://doi.org/10.3390/plants9040543
Li J, Xie D-F, Guo X-L, Zheng Z-Y, He X-J, Zhou S-D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants. 2020; 9(4):543. https://doi.org/10.3390/plants9040543
Chicago/Turabian StyleLi, Jun, Deng-Feng Xie, Xian-Lin Guo, Zhen-Ying Zheng, Xing-Jin He, and Song-Dong Zhou. 2020. "Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship" Plants 9, no. 4: 543. https://doi.org/10.3390/plants9040543
APA StyleLi, J., Xie, D.-F., Guo, X.-L., Zheng, Z.-Y., He, X.-J., & Zhou, S.-D. (2020). Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants, 9(4), 543. https://doi.org/10.3390/plants9040543