The First Glimpse of Streptocarpus ionanthus (Gesneriaceae) Phylogenomics: Analysis of Five Subspecies’ Chloroplast Genomes

 ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Results
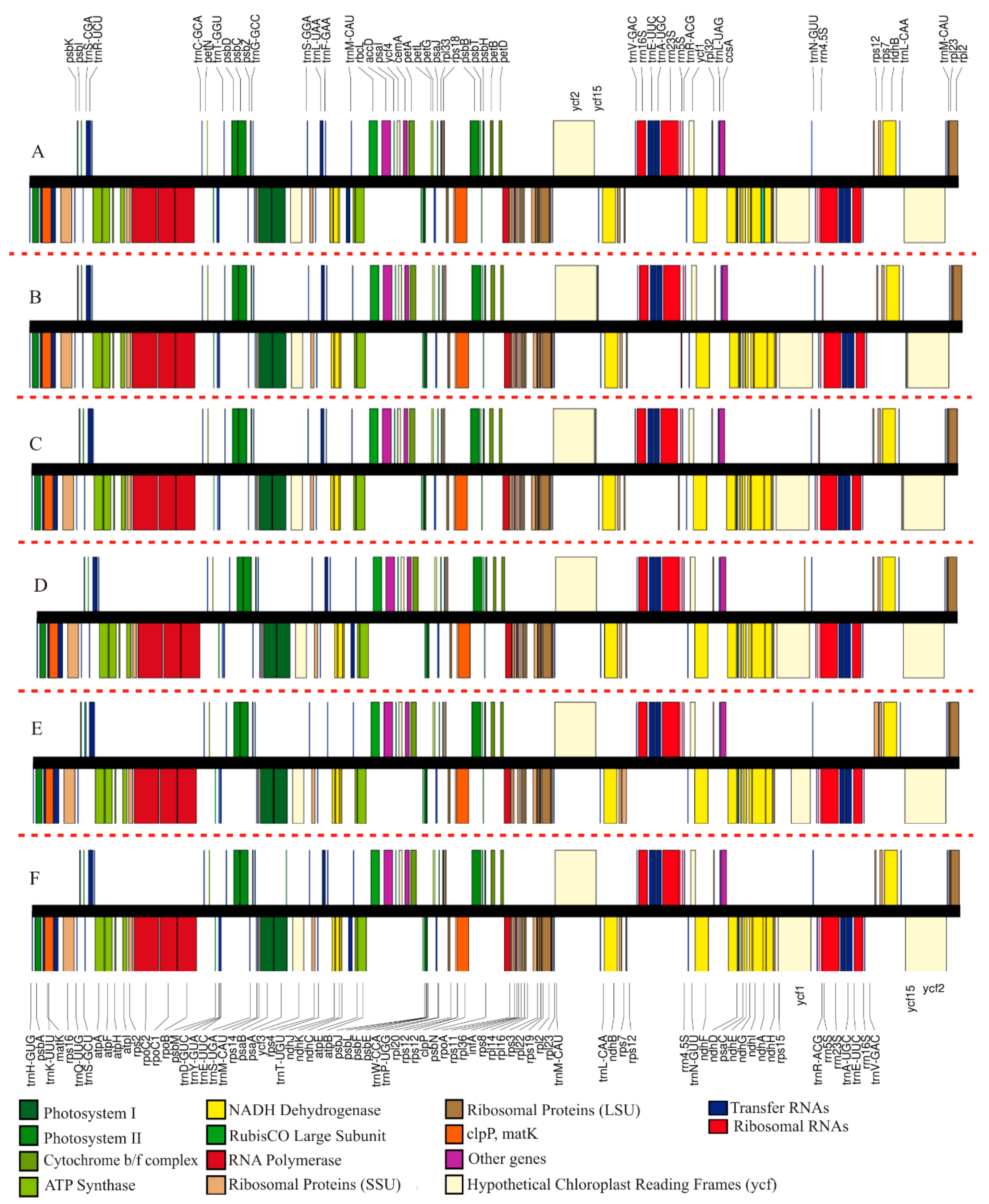
2.1. Overall Features of Str. ionanthus Chloroplast Genome
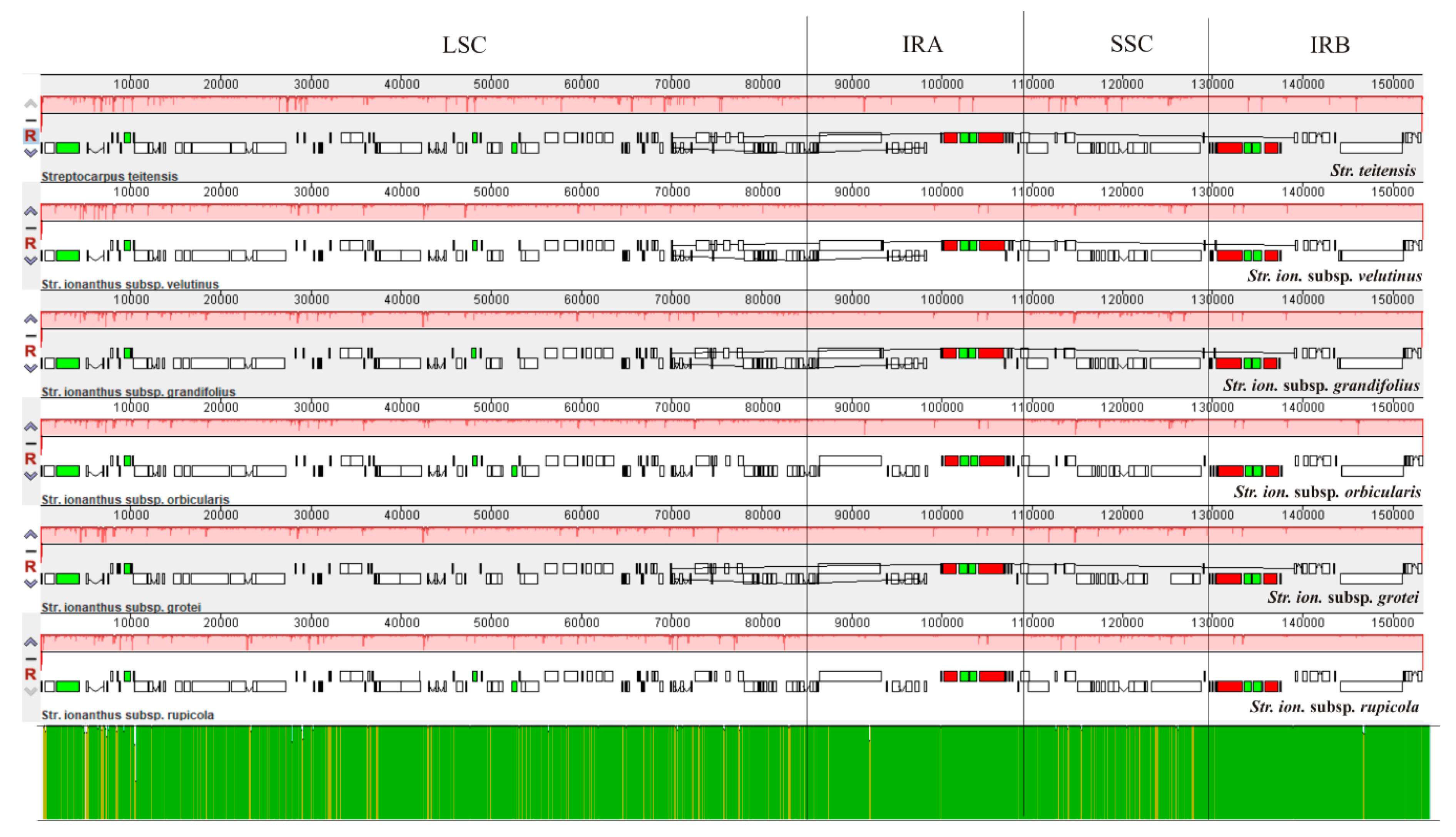
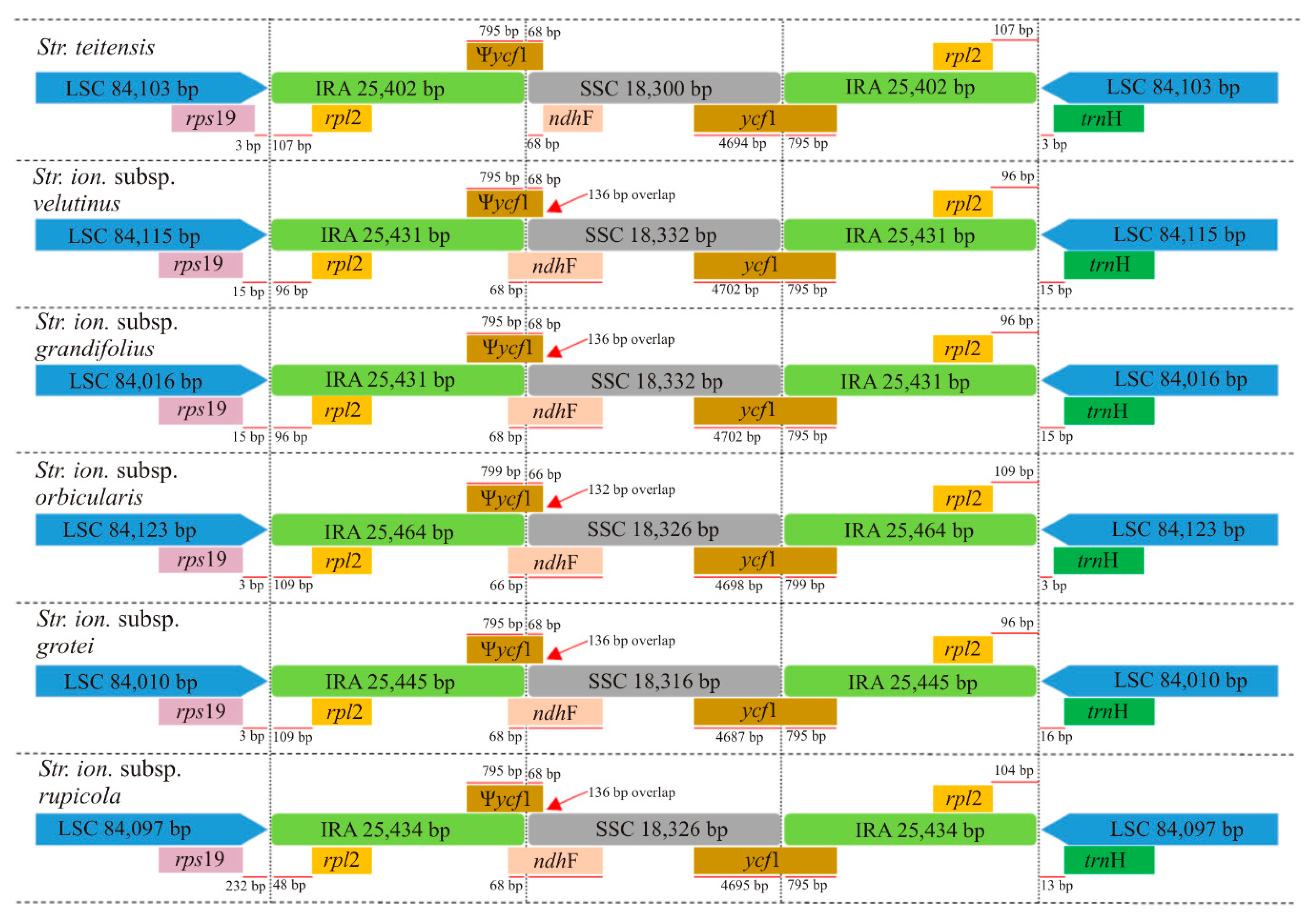
2.2. Comparison of Chloroplast Genome Structure in Sect. Saintpaulia
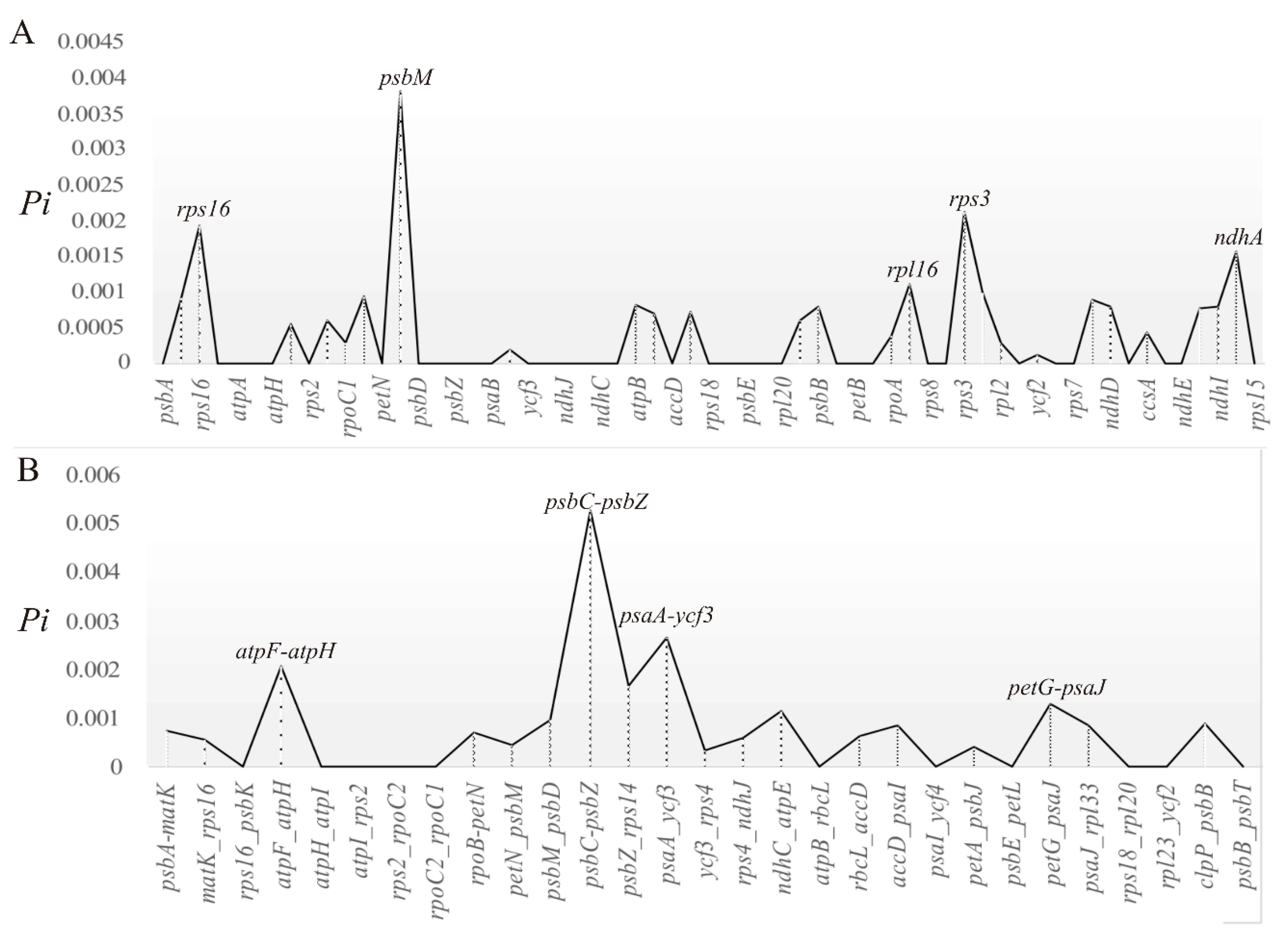
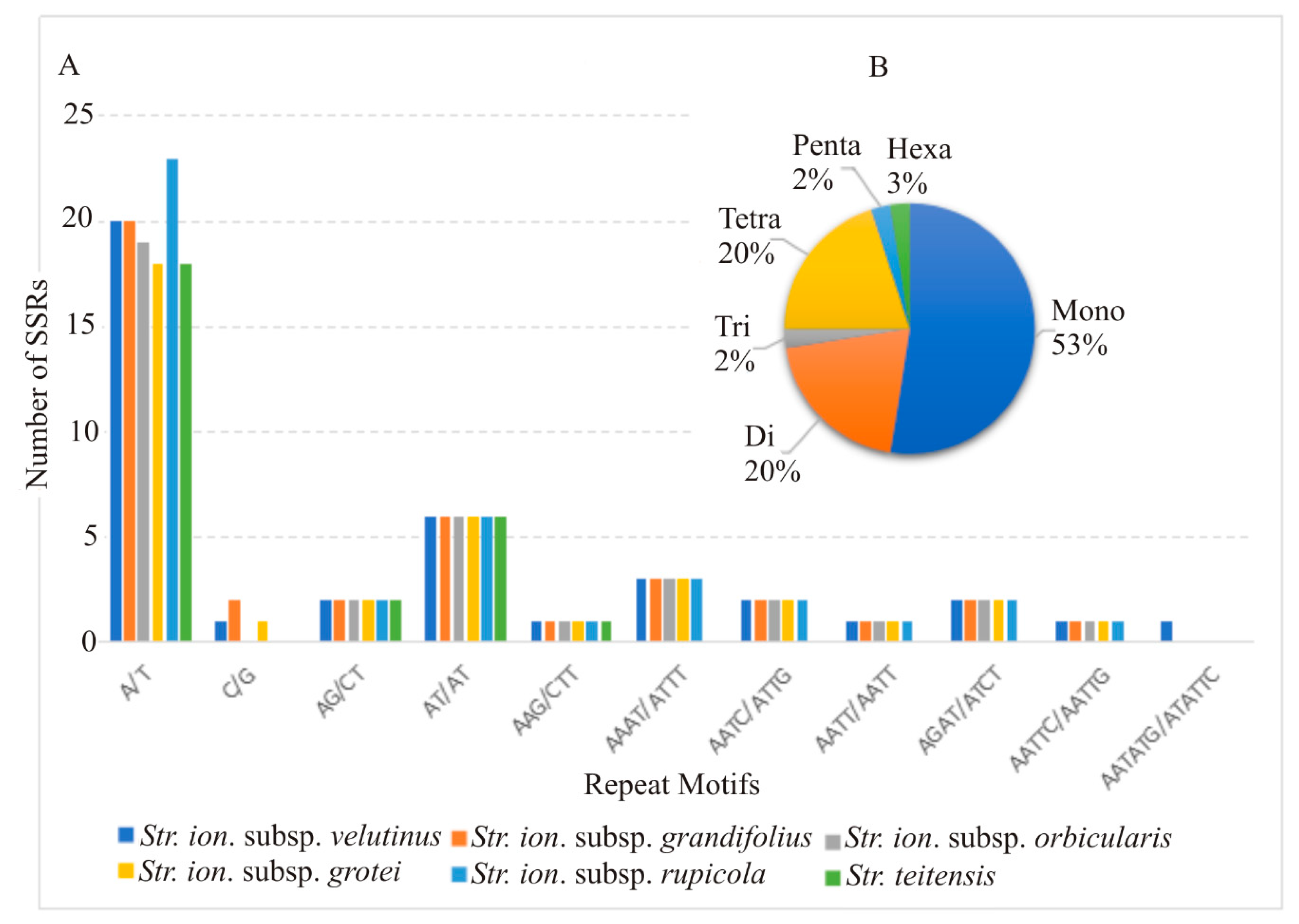
2.3. Divergent Hotspots and Simple Sequence Repeats (SSRs) in Str. ionanthus
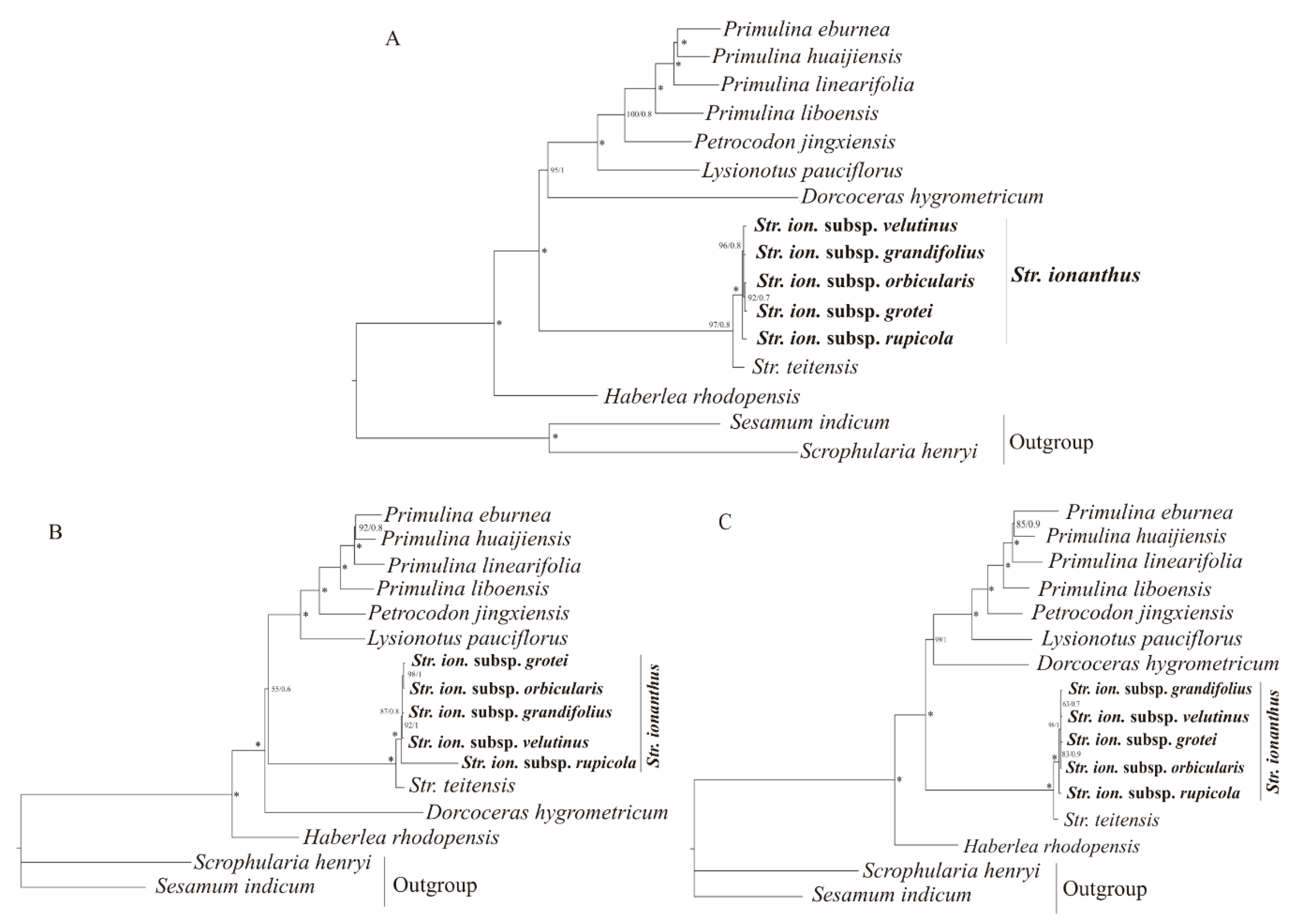
2.4. Phylogenetic Analysis
3. Discussion
3.1. Analysis of Genome Features
3.2. Divergence Hotspots in Str. ionanthus
3.3. Phylogenetic Relationship within Str. ionanthus
4. Materials and Methods
4.1. Sampling, Laboratory Experiments and Sequencing
4.2. Assembly and Gene Annotation
4.3. Genome Comparison
4.4. Identification of Divergent Hotspots and Simple Sequence Repeats (SSRs)
4.5. Phylogenetic Analysis
5. Perspectives on Streptocarpus ionanthus Research
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nishii, K.; Hughes, M.; Briggs, M.; Haston, E.; Christie, F.; DeVilliers, M.J.; Hanekom, T.; Roos, W.G.; Bellstedt, D.U.; Möller, M. Streptocarpus redefined to include all Afro-Malagasy Gesneriaceae: Molecular phylogenies prove congruent with geographical distribution and basic chromosome numbers and uncover remarkable morphological homoplasies. Taxon 2015, 64, 1243–1274. [Google Scholar] [CrossRef]
- Afkhami-Sarvestani, R.; Serek, M.; Winkelmann, T. Interspecific crosses within the Streptocarpus subgenus Streptocarpella and intergeneric crosses between Streptocarpella and Saintpaulia ionantha genotypes. Sci. Hortic. 2012, 148, 215–222. [Google Scholar] [CrossRef]
- Kolehmainen, J.; Korpelainen, H. Morphotypes, varieties, or subspecies?: Genetic diversity and differentiation of four Saintpaulia (Gesneriaceae) morphotypes from the East Usambara Mountains, Tanzania. Bot. J. Linn. Soc. 2008, 157, 347–355. [Google Scholar] [CrossRef]
- Dimitrov, D.; Nogues-Bravo, D.; Scharff, N. Why do tropical mountains support exceptionally high biodiversity? The Eastern Arc mountains and the drivers of Saintpaulia diversity. PLoS ONE 2012, 7, e48908. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.J. Pollination Observations of the African Violet In the Taita Hills, Kenya. J. East Afr. Nat. Hist. 2008, 97, 33–42. [Google Scholar] [CrossRef]
- IUCN SSC East African Plants Red List Authority 2014. Saintpaulia ionantha. The IUCN Red List of Threatened Species 2014: e.T158153A763135. https://dx.doi.org/10.2305/IUCN.UK.2014-1.RLTS.T158153A763135.en. Downloaded on 07 April 2020. Available online: https://www.iucnredlist.org/species/158153/763135 (accessed on 3 April 2020).
- Moller, M.; Cronk, Q.C. Phylogeny and disjunct distribution: Evolution of Saintpaulia (Gesneriaceae). Proc. Biol. Sci 1997, 264, 1827–1836. [Google Scholar] [CrossRef]
- Moller, M.; Cronk, Q.C.B. Origin and relationships of Saintpaulia (Gesneriaceae) based on Ribosomal DNA Internal Transcribed Spacer (ITS) sequences. Am. J. Bot. 1997, 84, 956–965. [Google Scholar] [CrossRef]
- Lindqvist, C.; Albert, V.A. A High Elevation Ancestry for the Usambara Mountains and Lowland Populations of African Violets (Saintpaulia, Gesneriaceae). Syst. Geogr. Plants 2001, 71, 37–44. [Google Scholar] [CrossRef]
- Burtt, B.L. Studies in the Gesneriaceae of the old world. XV. The genus Saintpaulia. R. Bot. Gard. Edinb. 1958, 22, 547–568. [Google Scholar]
- Burtt, B.L. Studies in the Gesneriaceae of the Old World 25: Additional notes on Saintpaulia. Notes R. Bot. Gard. Edinb. 1964, 25, 191–195. [Google Scholar]
- Darbyshire, I. Gesneriaceae. In Flora of Tropical East Africa; Beentje, H.J., Ghazanfar, S.A., Eds.; Royal Botanic Gardens: Kew, UK, 2006. [Google Scholar]
- Qiao, J.; Cai, M.; Yan, G.; Wang, N.; Li, F.; Chen, B.; Gao, G.; Xu, K.; Li, J.; Wu, X. High-throughput multiplex cpDNA resequencing clarifies the genetic diversity and genetic relationships among Brassica napus, Brassica rapa and Brassica oleracea. Plant Biotechnol. J. 2016, 14, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Y.; Liu, Y.-M.; Xie, D.-F.; He, X.-J.; Zhou, S.-D. Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution. Plants 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Lv, J.; Xu, J.; Zhu, S.; Li, M. Comparative Chloroplast Genomes of Sorghum Species: Sequence Divergence and Phylogenetic Relationships. Biomed Res. Int. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Souza, U.J.; Nunes, R.; Targueta, C.P.; Diniz-Filho, J.A.; de Campos Telles, M.P. The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae - caesalpinioideae): Comparative analysis with related Mimosoid species. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef]
- Cai, J.; Ma, P.F.; Li, H.T.; Li, D.Z. Complete Plastid Genome Sequencing of Four Tilia Species (Malvaceae): A Comparative Analysis and Phylogenetic Implications. PLoS ONE 2015, 10, e0142705. [Google Scholar] [CrossRef]
- Pyron, R.A.; Hendry, C.R.; Chou, V.M.; Lemmon, E.M.; Lemmon, A.R.; Burbrink, F.T. Effectiveness of phylogenomic data and coalescent species-tree methods for resolving difficult nodes in the phylogeny of advanced snakes (Serpentes: Caenophidia). Mol. Phylogenetics Evol. 2014, 81, 221–231. [Google Scholar] [CrossRef]
- Mitchell, N.; Lewis, P.O.; Lemmon, E.M.; Lemmon, A.R.; Holsinger, K.E. Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L. Am. J. Bot. 2017, 104, 102–115. [Google Scholar] [CrossRef]
- Kyalo, C.M.; Gichira, A.W.; Li, Z.Z.; Saina, J.K.; Malombe, I.; Hu, G.W.; Wang, Q.F. Characterization and Comparative Analysis of the Complete Chloroplast Genome of the Critically Endangered Species Streptocarpus teitensis (Gesneriaceae). Biomed. Res. Int. 2018, 2018, 1507847. [Google Scholar] [CrossRef]
- Lee, S.-R.; Kim, K.; Lee, B.-Y.; Lim, C.E. Complete chloroplast genomes of all six Hosta species occurring in Korea: Molecular structures, comparative, and phylogenetic analyses. BMC Genom. 2019, 20, 833. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Li, L.; Gao, L.; Xu, J.; Yang, M. Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species. Int. J. Mol. Sci. 2018, 10, 3262. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Guo, X.; Liu, Q.; Wang, K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 2019, 14, e0216645. [Google Scholar] [CrossRef] [PubMed]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, Z.; Sablok, G.; Daskalova, E.; Zahmanova, G.; Apostolova, E.; Yahubyan, G.; Baev, V. Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response. Front. Plant Sci. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.-B.; Fu, L.-F.; Fu, Z.-X.; Li, S.; Wei, Y.-G.; Wen, F. Complete chloroplast genome sequence of Petrocodon jingxiensis (Gesneriaceae). Mitochondrial DNA Part B 2019, 4, 2771–2772. [Google Scholar] [CrossRef]
- Feng, C.; Xu, M.; Feng, C.; von Wettberg, E.J.B.; Kang, M. The complete chloroplast genome of Primulina and two novel strategies for development of high polymorphic loci for population genetic and phylogenetic studies. BMC Evol. Biol. 2017, 17, 224. [Google Scholar] [CrossRef]
- Badger, J.H.; Zhang, T.; Fang, Y.; Wang, X.; Deng, X.; Zhang, X.; Hu, S.; Yu, J. The Complete Chloroplast and Mitochondrial Genome Sequences of Boea hygrometrica: Insights into the Evolution of Plant Organellar Genomes. PLoS ONE 2012, 7, e30531. [Google Scholar]
- Ren, T.; Zheng, W.; Han, K.; Zeng, S.; Zhao, J.; Liu, Z.-L. Characterization of the complete chloroplast genome sequence of Lysionotus pauciflorus (Gesneriaceae). Conserv. Genet. Resour. 2016, 9, 185–187. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.; Yan, C.; Zhao, X.; Shan, S. A comparative analysis of the complete chloroplast genome sequences of four peanut botanical varieties. PeerJ 2018, 6, e5349. [Google Scholar] [CrossRef]
- Fan, W.B.; Wu, Y.; Yang, J.; Shahzad, K.; Li, Z.H. Comparative Chloroplast Genomics of Dipsacales Species: Insights Into Sequence Variation, Adaptive Evolution, and Phylogenetic Relationships. Front. Plant Sci. 2018, 9, 689. [Google Scholar] [CrossRef]
- Liu, H.; He, J.; Ding, C.; Lyu, R.; Pei, L.; Cheng, J.; Xie, L. Comparative Analysis of Complete Chloroplast Genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica Revealing Structural Variations Among Genera in Tribe Anemoneae (Ranunculaceae). Front. Plant Sci. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.F.; Li, Z.H. Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 2019, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Zheng, C.; Tian, X.; Zheng, M.; Xiang, B.; Wang, Y. Comparison of Four Complete Chloroplast Genomes of Medicinal and Ornamental Meconopsis Species: Genome Organization and Species Discrimination. Sci. Rep. 2019, 9, 10567. [Google Scholar] [CrossRef]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin, A.W.T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Chung, H.Y.; Won, S.Y.; Kim, Y.-K.; Kim, J.S. Development of the chloroplast genome-based InDel markers in Niitaka (Pyrus pyrifolia) and its application. Plant Biotechnol. Rep. 2019, 13, 51–61. [Google Scholar] [CrossRef]
- Cho, M.S.; Kim, J.H.; Kim, C.S.; Mejías, J.A.; Kim, S.C. Sow Thistle Chloroplast Genomes: Insights into the Plastome Evolution and Relationship of Two Weedy Species, Sonchus asper and Sonchus oleraceus (Asteraceae). Genes 2019, 10, 181. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y.; Song, J.; Yao, H. Molecular Structure and Phylogenetic Analyses of Complete Chloroplast Genomes of Two Aristolochia Medicinal Species. Int. J. Mol. Sci. 2017, 18, 1829. [Google Scholar] [CrossRef]
- Lu, R.-S.; Li, P.; Qiu, Y.-X. The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 2016, 7, 2054. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, Y.; Chen, X.; Li, Y.; Xu, Z.; Duan, B.; Li, Y.; Song, J.; Yao, H. Complete Chloroplast Genomes of Papaver rhoeas and Papaver orientale: Molecular Structures, Comparative Analysis, and Phylogenetic Analysis. Molecules 2018, 23, 437. [Google Scholar] [CrossRef]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Chung, M.G.; Park, S. The Complete Chloroplast Genome Sequences of Three Veroniceae Species (Plantaginaceae): Comparative Analysis and Highly Divergent Regions. Front. Plant Sci. 2016, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Ngo Ngwe, M.F.; Omokolo, D.N.; Joly, S. Evolution and Phylogenetic Diversity of Yam Species (Dioscorea spp.): Implication for Conservation and Agricultural Practices. PLoS ONE 2015, 10, e0145364. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.; Rousseau-Gueutin, M.; Martin, G.E.; Morice, J.; Boutte, J.; Coissac, E.; Ourari, M.; Aïnouche, M.; Salmon, A.; Cabello-Hurtado, F.; et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 2017, 24, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lu, Y.; Xie, X.; Li, B.; Han, Y.; Sun, T.; Xian, Y.; Yang, H.; Liu, K. Development of chloroplast genomic resources for Pyrus hopeiensis (Rosaceae). Conserv. Genet. Resour. 2017, 10, 511–513. [Google Scholar] [CrossRef]
- Knowles, L.L.; Chan, Y.-H. Resolving Species Phylogenies of Recent Evolutionary Radiations. Ann. Mo. Bot. Gard. 2008, 95, 224–231. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 4, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Yang, L.; Yang, Z.; Liu, C.; He, Z.; Zhang, Z.; Yang, J.; Liu, H.; Yang, J.; Ji, Y. Chloroplast phylogenomic analysis provides insights into the evolution of the largest eukaryotic genome holder, Paris japonica (Melanthiaceae). BMC Plant Biol. 2019, 19, 293. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Beier, S.; Thomas, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Bui, Q.M.; Wong, T.K.F.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Str. teitensis | Str. ionanthus subsp. velutinus | Str. ionanthus subsp. grandifolius | Str. ionanthus subsp. orbicularis | Str. ionanthus subsp. grotei | Str. ionanthus subsp. rupicola |
|---|---|---|---|---|---|---|
| Accession Number | MF596485 | MN935472 | MN935471 | MN935470 | MN935469 | MN935473 |
| Total size (bp) | 153,207 | 153,307 | 153,208 | 153,377 | 153,215 | 153,290 |
| LSC size (bp) | 84,103 | 84,115 | 84,016 | 84,123 | 84,010 | 84,097 |
| SSC size (bp) | 18,300 | 18,332 | 18,332 | 18,326 | 18,316 | 18,326 |
| IR size (bp) | 25,402 | 25,431 | 25,431 | 25,464 | 25,445 | 25,434 |
| Number of genes | 114 | 115 | 115 | 115 | 115 | 115 |
| Number of PCGs | 79 | 80 | 80 | 80 | 80 | 80 |
| Number of tRNAs | 31 | 31 | 31 | 31 | 31 | 31 |
| Number of rRNAs | 4 | 4 | 4 | 4 | 4 | 4 |
| Category | Gene Names |
|---|---|
| Photosystem 1 | psaA, psaB, psaC, psaI, psaJ |
| Photosystem 11 | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| NADH Dehydrogenase | ndhA a, ndhB a,c, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| ATP Synthase | atpA, atpB, atpE, atpF a, atpH, atpI |
| Cytochrome b/f complex | petA, petB, petD, petG, petL, petN |
| RubisCO large subunit | rbcL |
| RNA Polymerase | rpoA, rpoB, rpoC1 a, rpoC2 |
| Ribosomal proteins (Large) | rpl2 a, rpl14, rpl16, rpl20, rpl22, rpl23 c, rpl32, rpl33, rpl36 |
| Ribosomal proteins (Small) | rps2, rps3, rps4, rps7 c, rps8, rps11, rps12 b,c,d, rps14, rps15, rps16 a, rps18, rps19 |
| Ribosomal RNAs | rrn4.5 c, rrn5 c, rrn16 c, rrn23 c |
| Transfer RNAs | trnA-UGC a,c, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC, trnH-GUG, |
| trnI-CAU c, trnI-GAU a,c, trnK-UUU, trnL-CAA c, trnL-UAA a, trnL-UAG, trnfM-CAU, | |
| trnN-GUU c, trnP-UGG, trnQ-UUG, trnR-ACG c, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnS-CGA | |
| trnT-GGU, trnT-UGU, trnV-GAC c, trnV-UAC a, trnW-CCA, trnY-GUA, trnM-CAU | |
| ycf1 c, ycf2 c, ycf3 b, ycf4, ycf15 a,c | |
| Protease | clpP b |
| Maturase | matK |
| Translational initiation factor | infA |
| Envelope membrane protein | cemA |
| Subunit of acetyl-CoA-carboxylase | accD |
| c-type cytochrome synthesis | ccsA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyalo, C.M.; Li, Z.-Z.; Mkala, E.M.; Malombe, I.; Hu, G.-W.; Wang, Q.-F. The First Glimpse of Streptocarpus ionanthus (Gesneriaceae) Phylogenomics: Analysis of Five Subspecies’ Chloroplast Genomes. Plants 2020, 9, 456. https://doi.org/10.3390/plants9040456
Kyalo CM, Li Z-Z, Mkala EM, Malombe I, Hu G-W, Wang Q-F. The First Glimpse of Streptocarpus ionanthus (Gesneriaceae) Phylogenomics: Analysis of Five Subspecies’ Chloroplast Genomes. Plants. 2020; 9(4):456. https://doi.org/10.3390/plants9040456
Chicago/Turabian StyleKyalo, Cornelius M., Zhi-Zhong Li, Elijah M. Mkala, Itambo Malombe, Guang-Wan Hu, and Qing-Feng Wang. 2020. "The First Glimpse of Streptocarpus ionanthus (Gesneriaceae) Phylogenomics: Analysis of Five Subspecies’ Chloroplast Genomes" Plants 9, no. 4: 456. https://doi.org/10.3390/plants9040456
APA StyleKyalo, C. M., Li, Z.-Z., Mkala, E. M., Malombe, I., Hu, G.-W., & Wang, Q.-F. (2020). The First Glimpse of Streptocarpus ionanthus (Gesneriaceae) Phylogenomics: Analysis of Five Subspecies’ Chloroplast Genomes. Plants, 9(4), 456. https://doi.org/10.3390/plants9040456