3.1. Mixed Microbial Activities
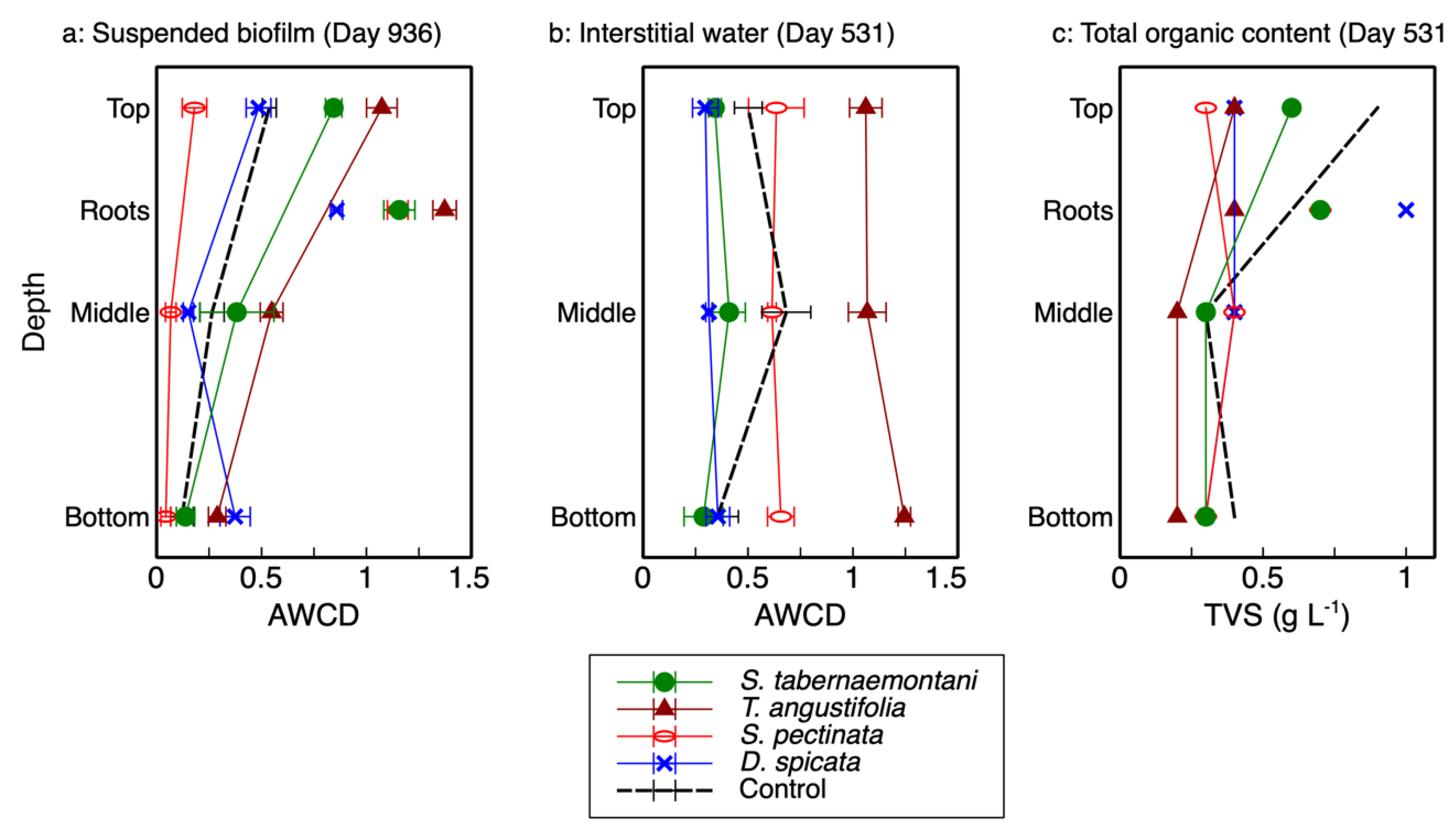
The AWCD—the average well color development in Biolog® plates and used as an indicator of microbial activity—and total richness for each reactor’s depth and plant root are presented in
Table 1. Unlike the previously reported microbial activity in the interstitial water samples [
2], which showed no consistent correlation with depth (
Figure 1b), the results from the wood-chip samples showed a clear decreasing trend as the reactor depth increased (
Figure 1a). As displayed in this figure, the activity of the microbial communities was highest at the top of each reactor (20-cm depth), decreased in the middle (40-cm depth) and, with the exception of
D. spicata, decreased further at the bottom (60-cm depth). The highest activity was recorded for the
T. angustifolia reactor, followed by
S. tabernaemontani. Moreover, the rhizospheric activities were higher than those associated with the reactor substrate, where the highest rhizospheric activity pertained to
T. angustifolia.
The two-way ANOVA on the wood-chip samples showed that both depth and vegetation were significant factors for the microbial activity (
p-value < 0.0001). The significantly different activity at each depth indicated a distinct stratification through the vertical profile of each reactor. The pair-wise comparisons showed the activities were also significantly different between all the reactors except for the
D. spicata and the unplanted units (
p-value = 0.9). According to the interaction effect of vegetation and depth, the reactor planted with
T. angustifolia showed significantly higher activities with respect to the control at all the three depths (
p-values < 0.005), whereas
S. tabernaemontani’s activities were significantly higher than control at the shallower depths (top and middle,
p-values < 0.05). These differences were, however, consistently less prominent as the depth increased (
Figure 1a).
The organic contents (TVS) of the detached biofilm from the top layer of
T. angustifolia,
S. tabernaemontani and control reactors were approximately two times higher than the middle and bottom layer samples (
Figure 1c). The higher organic mass detached from the top layers of these reactors suggest that, compared to the lower layers, a more substantial biofilm was formed at the shallowest depth; thus, the higher observed activities of the better-established communities in the upper layers of these reactors were consistent with the biofilm mass.
The total richness (
Table 1) showed a similar trend to the AWCD values, with a generally decreasing richness as the depth increased. Moreover, richness was closely correlated with AWCD values for wood-chip samples, as richness was lower for smaller catabolic activities. The declining richness in depth demonstrated a decrease in functional utility of the microbial community at greater depths [
7]. This observation is in agreement with other studies (e.g., [
7,
9]).
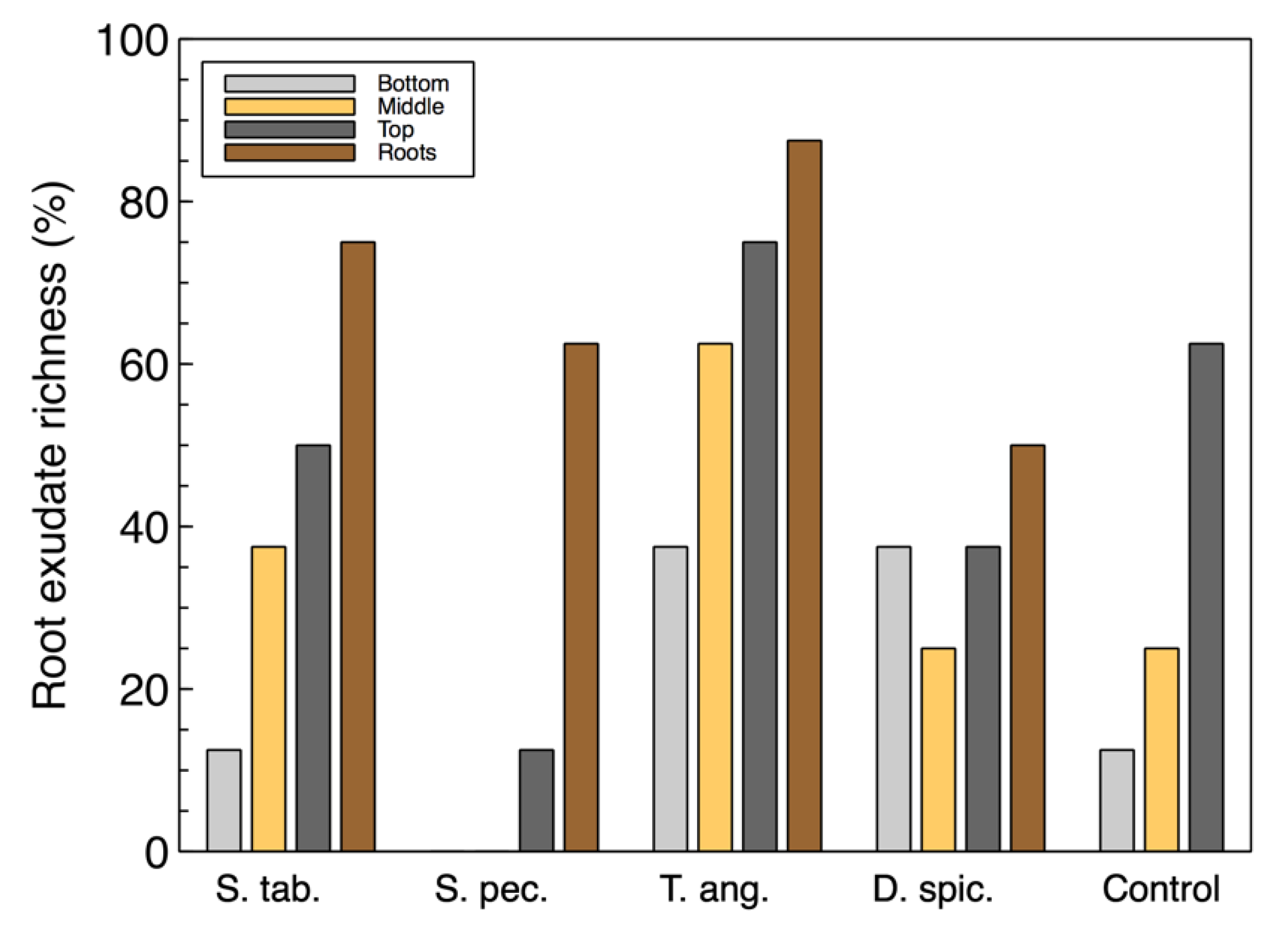
For the most part, the richness of the utilized carbon sources associated with root exudates (
Figure 2) were also similar to the total richness. However, the root exudate richness in the wood-chip samples from the unplanted reactor was found to be higher than some of the vegetated reactors. This suggests that although the control unit did not contain any plants, the microbial community was able to utilize these carbon sources and showed a richness of approximately 60%. This observation may also suggest that there is a link between the organisms capable of consuming root exudate sources and the colonized denitrifiers that were previously identified in the
T. angustifolia and control units [
2].
The higher activity in the reactors’ top layers and the greater rhizospheric activities compared to the activities associated with the wood chips alone were similar to the findings reported in treatment wetlands by Weber and Legge [
7]. Zhao et al. [
25] and Zhang et al. [
26] also found that plants had a strong effect on the diversity and activity of the microbial communities in CWs. This greater microbial activity is likely due to the presence of the plant roots, which were established at a higher density in the top layer of the reactors. The roots may have carried over external microbial sources to the reactors at the time of plantation. The other important contribution of roots is their physical structure, which is adept at fostering microbial communities.
Additionally, the exudates from the plant roots are known to play an important role in the rhizosphere biology through the interactions with the soil and the microbial community [
27]. Salomo et al. [
9] reported that the decayed plant litter in the surface layer of their CW could be utilized by the microorganisms present in that layer. The authors also suggested that a lack of nutrient supply in deeper layers may have an impact on the carbon source utilization pattern of the microbial communities. In our system, however, the catabolic activities of the
T. angustifolia and control reactors did not seem to suffer from the nitrate limitation that occurred in those units. Therefore, the combination of the potential external sources of microbes and physiochemical support of the root network, as well as the root exudates and presence of the plant litter were likely the main factors responsible for the higher activities and greater total richness in the
T. angustifolia and
S. tabernaemontani units.
It should, however, be noted that, compared with the unplanted unit, not all the plant species had a positive effect by increasing the microbial activity. The activity and richness of D. spicata and S. pectinata were often smaller than the control unit. These results suggest that the selection of a suitable plant species is an important factor for successful microbial development and growth and reactor treatment performance.
The AWCD of the biofilm can be compared with the AWCD of the interstitial water samples (
Figure 1a,b). In general, the microbial activities of the top layers in the
T. angustifolia and control reactors in the detached biofilm samples were similar to the average activities of the interstitial water samples in these two reactors. The other units did not show a consistent correlation between the two sampling methods. The biofilm extracted from the wood-chip substrate was able to provide a better picture of the developed stratification in the reactors. While the destructive sampling for biofilm extraction was conducted 405 days after the collection of interstitial water samples, the high microbial activities and denitrification rates implied that the
T. angustifolia reactor had already acclimatized by the time water sampling was conducted. This indicates the stratified pattern of the microbial activities through depth was also likely present in the reactors when water sampling was performed. Thus, it was the interstitial water sampling method that failed to show the reactor’s stratification that was evident in the biofilm analysis.
The lack of a recognizable trend from the water sampling method was likely due to the fact that some gravity-driven mixing of the vertical water flows was expected at the time of sampling. This could prevent each sample from being a perfect representation of the stagnant water phase adjacent to the biofilm. Furthermore, due to the unknown spacing between the tip of the sampling tubes buried inside the media and the coarse wood-chip grains, the characteristics of the collected water samples could differ between the sampling locations in one reactor and across all the reactors. That is, the buried end of one internal sampling tube could be in direct contact with a wood-chip grain, while another tube could be positioned a few millimeters away from the surface of a wood chip where biofilm would be formed. Therefore, the interstitial water sampling method did not seem to demonstrate the differences between the microbial activities along the reactors’ depth as well as the biofilm sampling did.
A total of five (
Figure S1a) and four (
Figure S1b) groups were identified based on the similarities of the data in multivariate analyses for the biofilm samples (PCA and AHC plots). The top layer sample of the wood-chip media in the
T. angustifolia reactor was grouped with all the plant roots, which is in agreement with the generally higher activities of these samples as reported in
Figure 1. The media samples in the top two layers of the
S. tabernaemontani reactor and the top layer of the unplanted and the middle layer of
T. angustifolia and control reactors were also grouped together. Although subtle, it seems that the similarity and dissimilarity of the biofilm samples are more influenced by different depths, rather than the planted species. This is in contrast to the similar analysis on the interstitial water samples, as shown in
Figure S2. In the interstitial water method, the grouping of the
T. angustifolia,
S. tabernaemontani and
D. spicata samples was more distinctly influenced by their respective plant species.
3.2. Denitrifying Gene Quantities
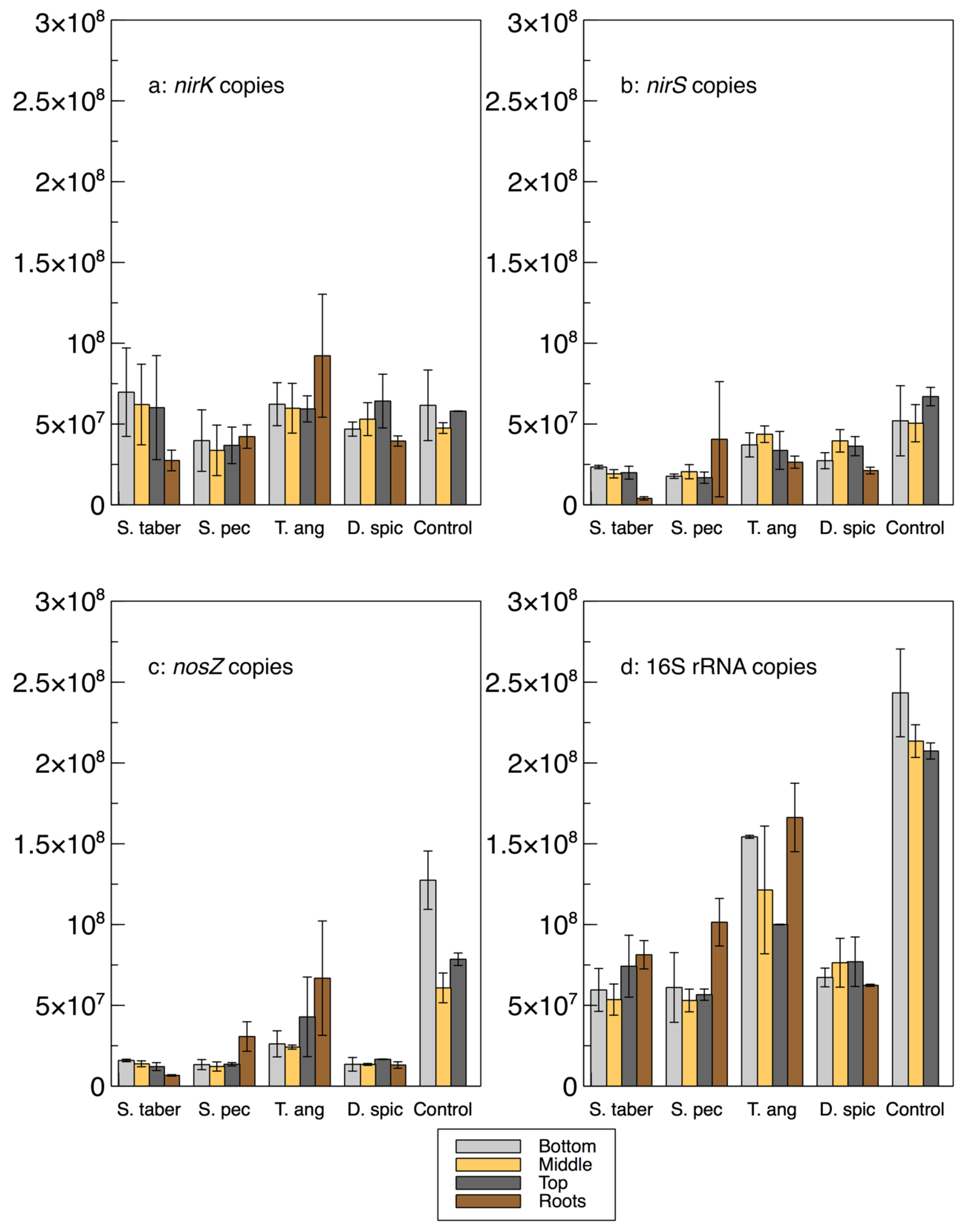
A relative qPCR method was used to quantify the abundance of the microbes encoding the
nirK,
nirS,
nosZ and 16S rRNA genes (
Figure 3 and
Table 2). As such, it is important to note that while the calculated gene copies provide important information about the bacterial communities and their abundance in relation to the other samples and genes, these values are not absolute numbers and, thus, cannot be directly compared with the gene copy numbers in the literature.
The abundance of the 16S rRNA gene was higher in the control and
T. angustifolia reactors, while
T. angustifolia’s roots showed the greatest abundance of 16S rRNA among the roots. Since the reactors were not augmented with an external source of bacteria, the total bacterial DNA (16S rRNA) abundance can be used as a general indicator for the natural development of the bacterial population in each reactor. The higher 16S rRNA count in these two reactors was in good agreement with their high denitrification performance at the time of the core sampling [
2]. Unlike the mixed community results (
Figure 1a), the 16S rRNA copies in these two reactors increased through the reactor depth, demonstrating the highest abundance in the bottom layer of the wood-chip media. The other reactors did not display a consistent pattern regarding their 16S rRNA copies at different depths. Except for the
D. spicata reactor, which was previously found to have the lowest microbial activity via CLPP (
Figure 1a), the abundance of 16S rRNA in the root samples of each reactor was higher than the reactor’s respective wood-chip samples, indicating a generally greater potential for the presence of the bacterial communities in the plants’ rhizosphere.
The average
nirK copies in the control reactor’s media was similar to the
nirS genes in this reactor, whereas
nirK copies were mostly higher than
nirS in the other units. The
nirK gene copies did not seem to reflect the nitrate removal performance of the reactors, shown in
Figure 4 and reported in Fatehi-Pouladi et al. [
2]. For instance, the
S. tabernaemontani reactor that showed a decreased denitrification efficiency towards the end of the experiment was found to be the unit with the highest
nirK copies. These results are in agreement with our earlier finding, where we reported the
nirK fold-changes in the reactors’ interstitial water were negligible [
2]. In the current paper, although
nirK was found in abundance and was typically greater than
nirS (
nirK/
nirS of between one to seven), its direct role in complete denitrification was less significant. This may suggest that the bacteria containing
nirK were present but may not have been expressing
nirK as much as the other nitrite reductase gene,
nirS, was expressed. As well, some of the denitrifying genes, including
nirK, are also present in bacteria involved in other biological nitrogen transformation pathways, such as incomplete denitrification and dissimilatory nitrate reduction to ammonia (DNRA) [
5].
The abundance of the
nirS gene was more consistent with the water quality results, as the control and
T. angustifolia reactors contained the highest
nirS counts, while the lowest
nirS abundance was found in the
S. tabernaemontani’s roots and wood-chip media. The differences between the
nirS gene copies among the reactors were, however, small compared to the
nosZ gene, whose average highest abundance in the control reactor was almost seven times higher than that of the lowest copies in the
S. pectinata unit. Although the cytochrome enzyme encoded by
nirS and the Cu-containing enzyme encoded by
nirK are needed to catalyze the reduction of nitrite to nitric oxide, the
nosZ enzyme is responsible for the last step of the denitrification pathway by reducing nitrous oxide [
28]. As such, the copy number of the bacteria with the
nosZ gene is an important indicator for a potentially complete denitrification pathway. The
nirS and
nosZ genes in our study appeared to be the key gene markers for the observed denitrification, whose populations were positively correlated with the nitrate reduction efficiencies.
Various proportions of the denitrifying genes have been reported in the literature. Warneke et al. [
29] found a
nirK/
nirS ratio of between 0.1 to 1.0 in their denitrification bed samples, and Ligi et al. [
24] reported a range of 0.07 to 0.74 for their wetland soils. These values were smaller than our ratios of 1.0 and up to 3.1 in the wood chips. Nonetheless, the reported
nosZ/
nir ratios of 0.1 to 0.7 [
29] and 0.04 to 0.40 [
24] were in agreement with our wood-chip
nosZ/
nir ratios of 0.2 to 0.8. The
nirK%,
nirS% and
nosZ% in Ligi et al. [
24] were generally much smaller than our values. However, given that the authors’ soil samples were from a river wetland area, a substantially different composition of bacterial community from that of our study is expected. Since our reactors were operated in a more isolated environment and were specifically designed to promote denitrification, it was anticipated that a higher ratio of the total bacterial population would express nitrite and nitrous oxide reductase genes. A similar observation was reported in Warneke et al. [
29], where
nirK%,
nirS% and
nosZ% ratios in multiple carbon-based denitrification barrels were higher than those reported in Ligi et al. [
24] for a potentially more complex wetland system.
Among the plant roots, T. angustifolia stood out by demonstrating the greatest copy numbers of nirK, nosZ, 16S rRNA, and nir+nosZ, as well as the ratios of nosZ% and nosZ/nir. These observations confirm the previously discussed important role of this species in the denitrifying bioreactors and further support that the maximum denitrification in this reactor, which occurred faster than the other reactors and benefited from its rhizosphere and its rich bacterial genome.
In general, and similar to the interstitial water samples, a consistent correlation between the denitrifying gene copies and reactor depth could not be established. Although the T. angustifolia and control reactors showed an inverse correlation between the 16S rRNA copies and the reactor depth, no consistent pattern could be identified for the other genes or in the other reactors. The decreased 16S rRNA copies in the T. angustifolia and control units in the lower depths could be due to the lack of substrates, particularly nitrate, in the layers closer to the reactor’s bottom.
Both interstitial water and biofilm sampling methods provided useful information regarding the potential activities and quantification of the denitrifying bacteria. The high gene abundance and gene fold-change in the reactors with high denitrification efficiency were recorded using the water and biofilm sampling methods, respectively. In the future, it is recommended to analyze these target genes using the absolute qPCR method, in which the standard curve is created by cloning known gene concentrations into plasmids. This method would further help to analyze the natural development of denitrifying bacteria and compare the expression levels with those of the other environmental samples.
3.3. 16S rRNA Amplicon Sequencing—Interstitial Water
The temporal changes of the bacterial composition in the interstitial water samples from the
S. tabernaemontani,
T. angustifolia and unplanted reactors are presented in
Figures SI3–SI6. In the
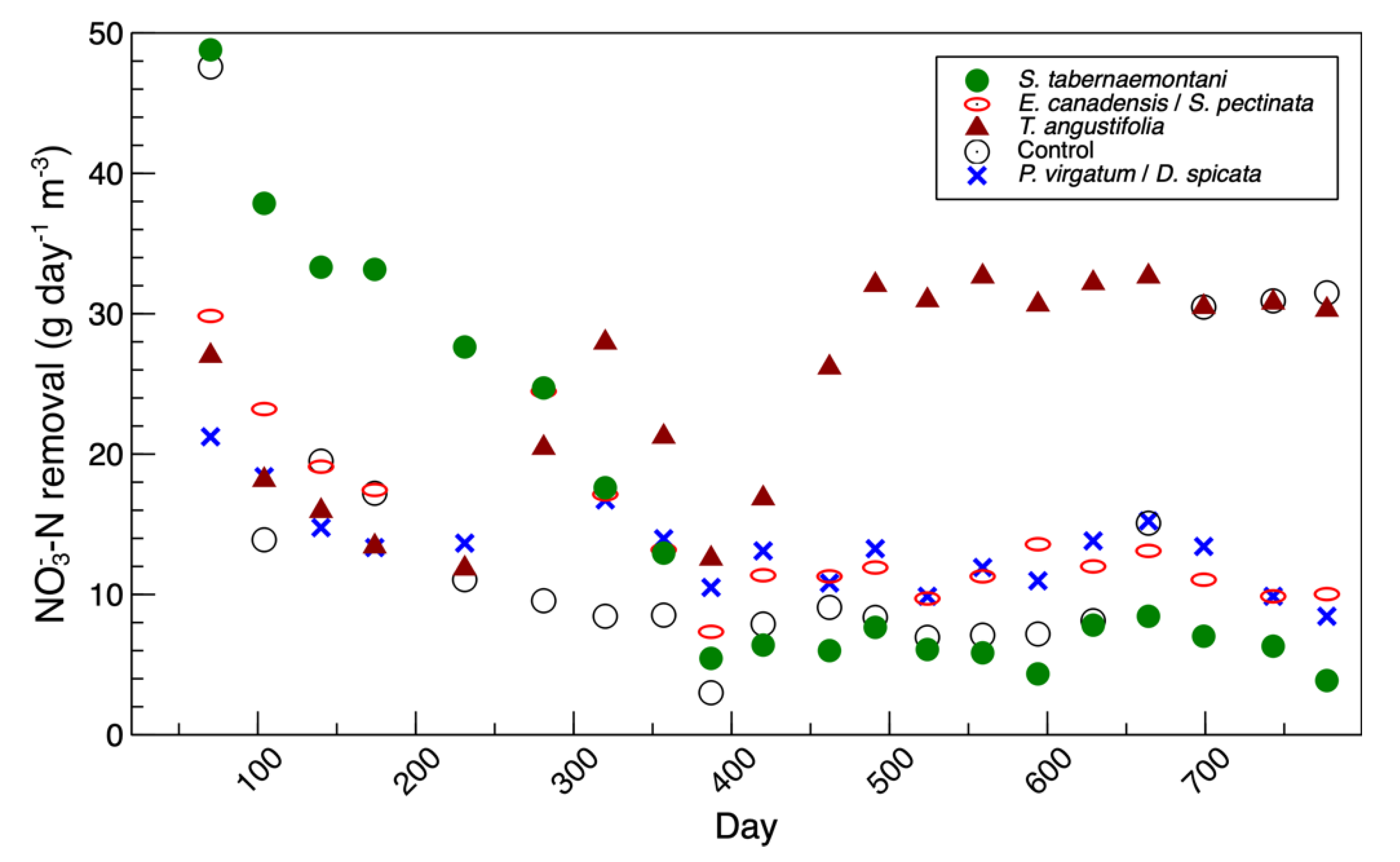
S. tabernaemontani reactor, the total abundance of the classified genera was highest in the beginning of the experiment (day 147,
Figure S3). This was consistent with the denitrification performance of this reactor, which was at its peak during the beginning phase of the study (
Figure 4). The
Bacillus genera had the highest abundance in this sampling event. It has been reported that denitrification is a common feature of
Bacillus spp. [
30], and the species related to this genus are among the most common denitrifying bacteria [
31]. The other abundant genera on day 147 were
Clostridium,
Leuconostoc,
Rhodococcus and
Longilinea, which were not greater than 2% during the later time points. The dominant classified genus in the rest of the water samples from
S. tabernaemontani was
Geobacillus, which had an abundance of less than 1% on day 147 but were consistently found in other sampling events (maximum of 13.3% on day 389). After
Geobacillus, the communities related to genera
Pseudoalteromonas and
Deinococcus were among the abundant groups between days 359 and 517, while
Weissella was the second-most abundant genus on the final sampling day (day 665). Compared to the samples collected approximately one year after the project start-up (days 359 to 665), the distinct structural composition of the water samples on day 147 indicated that these different bacterial communities played an important role in the denitrification that occurred early in this reactor.
A clear shift in the bacterial composition of the water samples in the
T. angustifolia reactor was found between days 442 and 517 (
Figure S4). This was consistent with the maximum nitrate reduction, which was achieved near day 500 (
Figure 4). Similar to the
S. tabernaemontani reactor,
Geobacillus was the most abundant genus in the
T. angustifolia reactor before day 516, but the major communities present in the second phase were
Sulfurimonas (max: 24.8%),
Sulfurospirillum (max: 8.0%),
Bacillus (max: 11.6%) and
Thauera (max: 18.6%). In addition, the final samples on day 665 contained abundant
Thermogemmatispora (max: 8.7%) and
Geobacter (max: 11.2%) spp.
Sulfurospirillum and
Thauera are both nitrate-reducing bacteria [
32,
33].
Sulfurimonas paralvinellae, one of the species prevalent in this reactor (
Figure S5), is a facultatively anaerobic chemolithoautotroph, sulfur- and thiosulfate-oxidizing bacteria, which can use nitrate or ammonium as its nitrogen source [
34]. The sulfate reduction that was observed in this reactor between days 500 and 700 (data not shown) can be explained by the sulfate-reducing bacteria (SRB), such as species affiliated with the genus
Desulfotomaculum [
35]. These organisms were found among the abundant species on day 601 (
Desulfotomaculum acetoxidans at approximately 2.3%) but decreased to an average of 0.8% on day 665. Nonetheless, the well-studied sulfate-reducing bacteria from the genus
Desulfovibrio and order
Desulfovibrionales [
36] were less than 0.37% during the entire operation.
The most abundant species in the water samples from the T. angustifolia reactor from day 517 onwards included Thauera aromatica (max: 12.6% on day 517), Bacillus ginsenggisoli (max: 8.6% on day 601), Sulfurimonas paralvinellae (max: 2.7% on day 517) and Thermogemmatispora foliorum and Geobacter lovleyi (max: 8.6% and 6.3% on day 665).
In the unplanted reactor, the colonies affiliated with the genus
Mycoplasma were present in most of the samples (max: 14.2%), although the ratio demonstrated a decreasing trend toward day 665 (
Figure S6).
The abundant species from this genus was
Mycoplasma insons, which was found between 0.2% and 13.2% (data not shown). The other main abundant genera in this reactor were
Geobacillus (max: 5.8%) and
Alkalibacterium (max: 5.1%). The
Geobacillus spp. are of interest in wood-chip bioreactors, since they comprise a group of bacteria that include denitrifiers and are known for their potential for degradation of hemicellulose and lignocellulose biomass, as well as their application in lignocellulosic biofuel production [
37,
38].
Bacillus spp. are similarly capable of utilizing alkaline lignin as a carbon or energy source [
39]. These species may have contributed to the breakdown of the organic matter from wood chips that was quantified by the high outflow five-day biochemical oxygen demand (BOD
5) and chemical oxygen demand (COD) in the
T. angustifolia and unplanted reactors (refer to [
2] for more details).
The influent sample was expectedly less diverse than the wood-chip bioreactors (
Figure S7). There were only two abundant genera with a ratio of greater than or equal to 2%, namely
Burkholderia (27%) and
Salinispora (31%), while the most abundant species was
Salinispora tropica (30.1%).
Salinispora spp. are the bacteria originating from a saline habitat and require seawater or sodium for growth [
40]. Thus, it is not surprising that these organisms were established in our synthetic greenhouse effluent with high salinity.
3.4. 16S rRNA Amplicon Sequencing—Biofilm
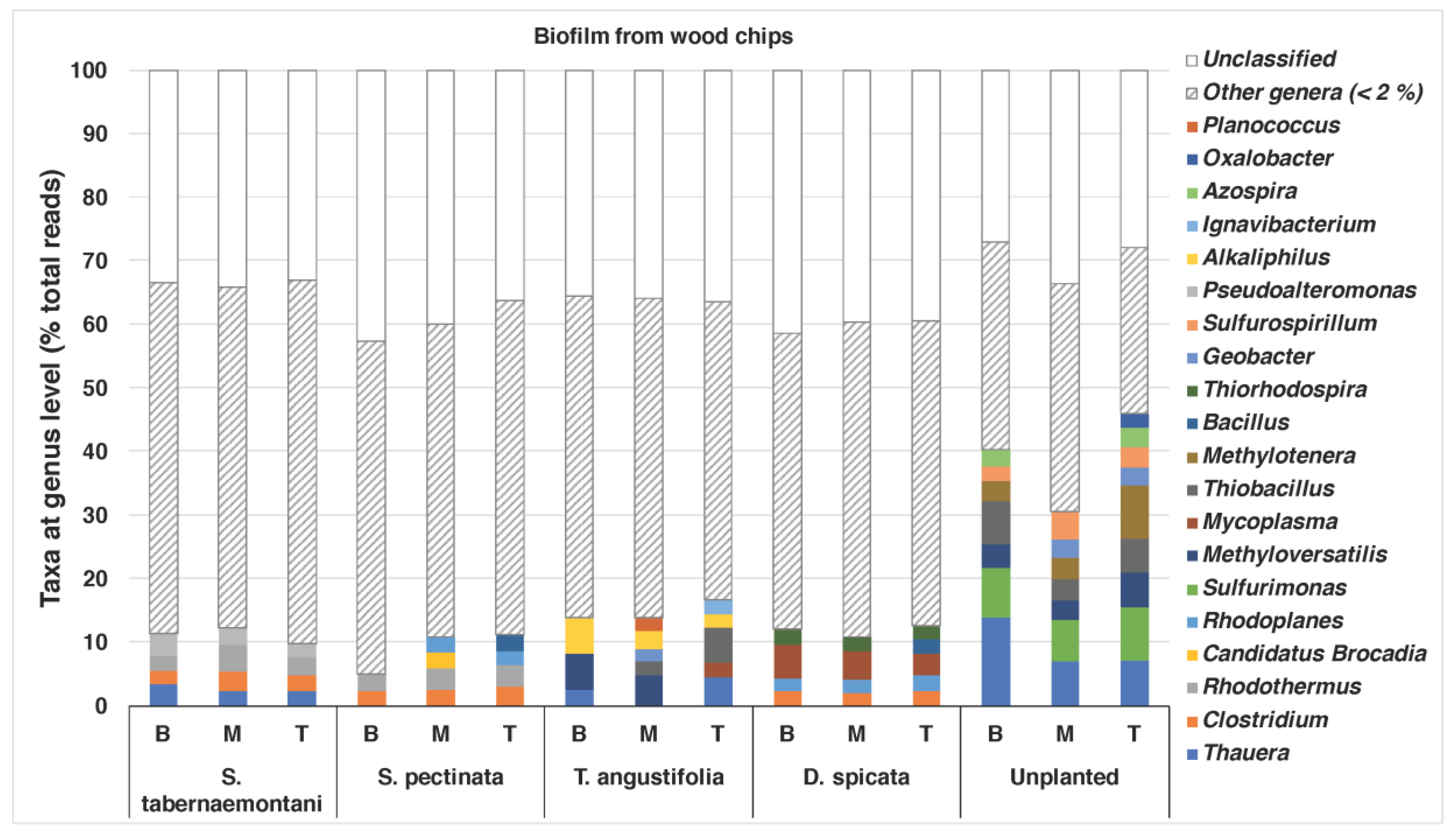
The bacterial compositions of the biofilm that was extracted from the wood chips are shown in
Figure 5. The unplanted reactor exhibited the most diverse structure of the genera, with abundances of >/= 2%. This reactor showed the highest nitrate and sulfate reductions among the other units near the end of the experiment when the core sampling was performed (data for sulfate reduction not shown).
Thauera (max: 13.9%),
Sulfurimonas (max: 8.3%),
Methyloversatilis (max: 5.6%),
Thiobacillus (max: 6.8%) and
Methylotenera (max: 8.4%) were among the identified genera in this reactor. The genera
Thauera,
Methyloversatilis and
Thiobacillus were also found in the wood chips from the
T. angustifolia reactor (max: 4.5%, 5.6% and 5.4%, respectively), which achieved a similar high denitrification and sulfate reduction earlier than the unplanted reactor.
Methyloversatilis has been shown to have denitrification capacity by reducing nitrate to nitrite [
41], and
Thauera spp. have been associated with denitrifying communities [
5]. The
T. angustifolia reactor was also the only unit that had abundant
Alkaliphilus spp., while the
S. tabernaemontani,
S. pectinata and
D. spicata had between 2% and 3% of the genus
Clostridium.
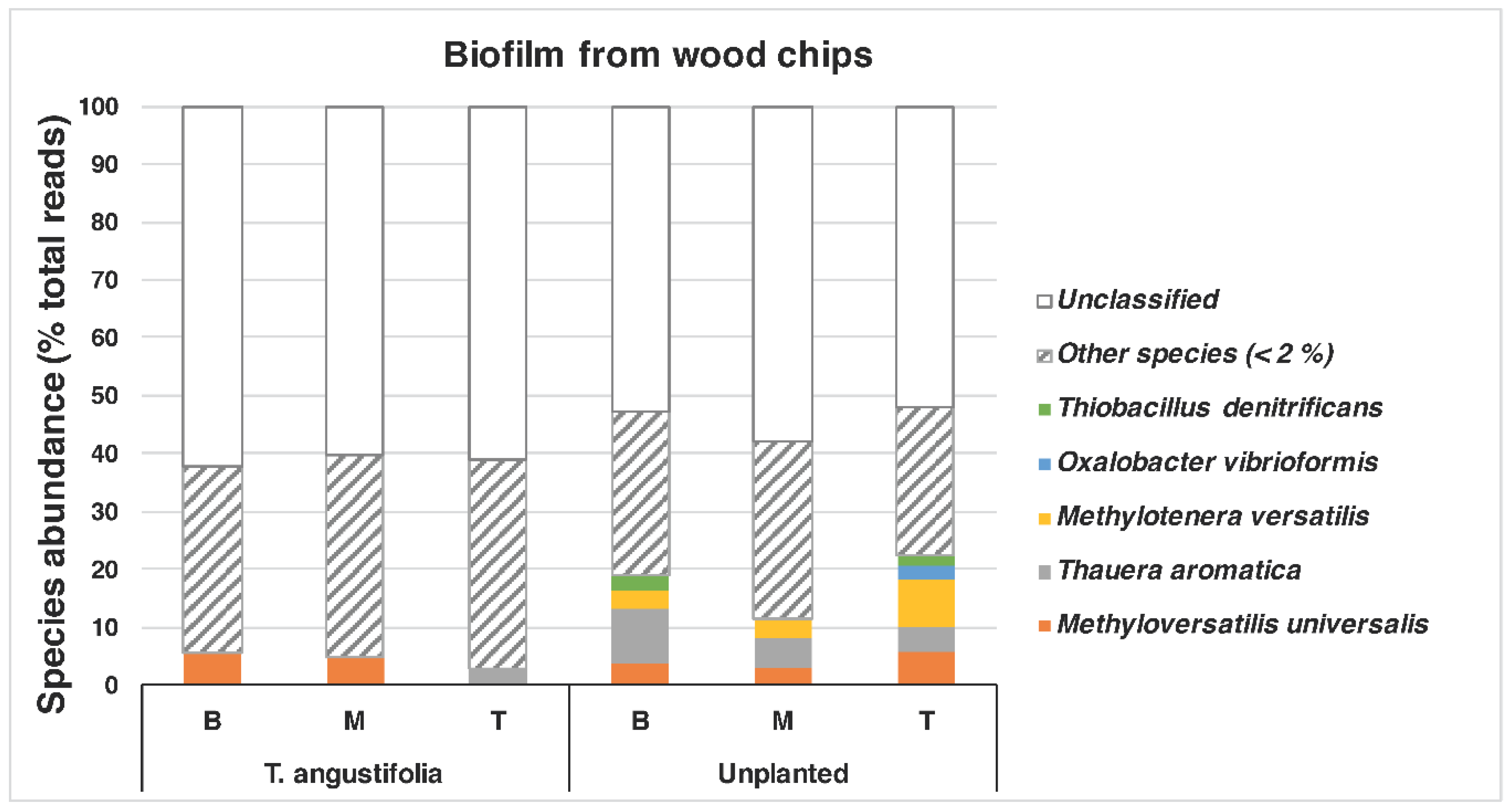
A closer look at the species found in the
T. angustifolia and unplanted reactors revealed that
Thauera aromatica (max: 9.5%) and
Methyloversatilis universalis (max: 5.6%) were among the dominant species in the media of these reactors, whereas
Methylotenera versatilis was mostly found only in the unplanted reactor (max: 8.4%) (
Figure 6).
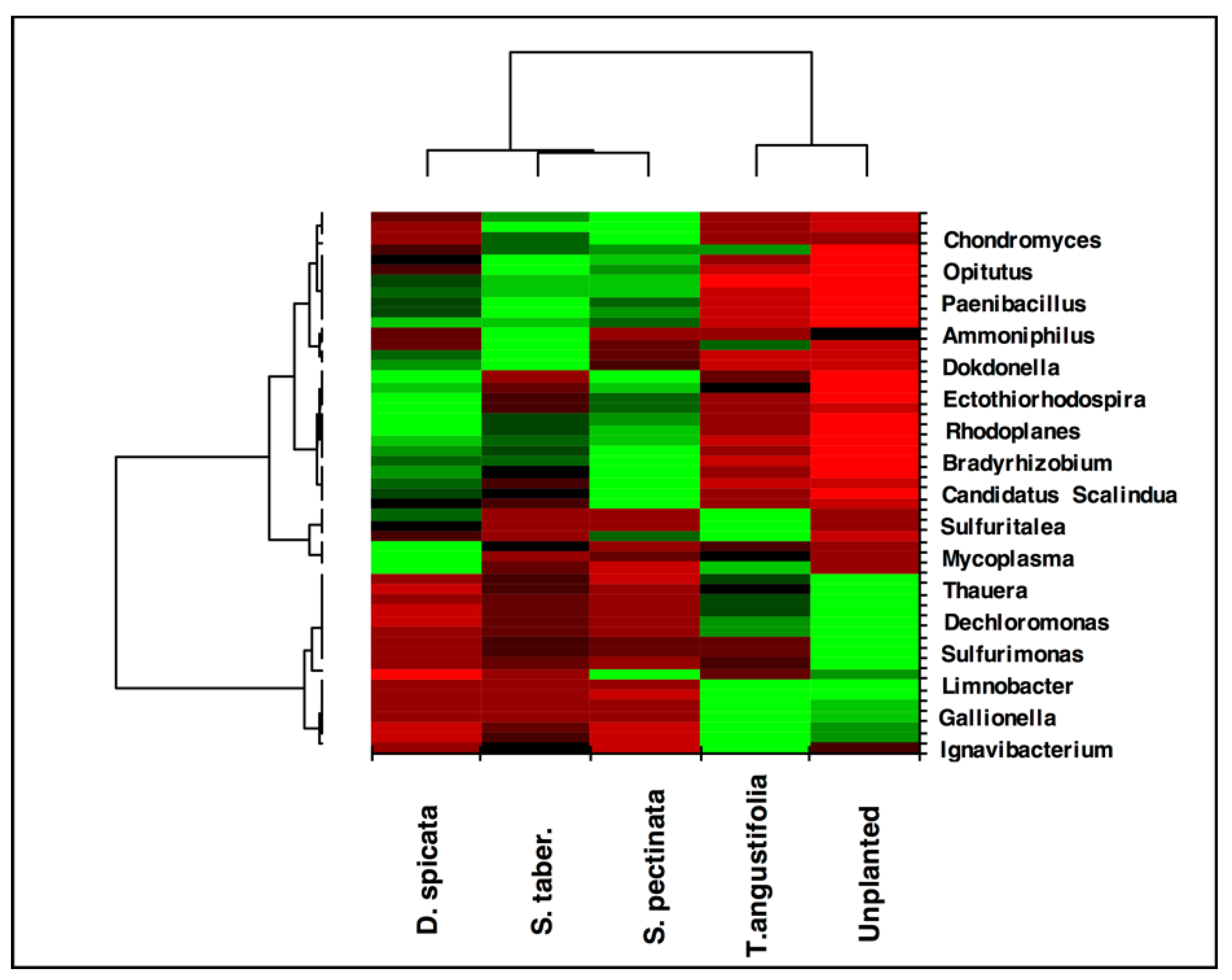
The heat map created for the taxa ratios at the genus level demonstrated high similarity between the
T. angustifolia and unplanted reactors (
Figure 7). This grouping indicates that these two reactors developed a generally similar microbial community at the end of the experiment, and the populations related to the genera found in higher abundance in these reactors were responsible for the similar treatment performance observed in these units.
The genera found in the root samples were generally less diverse compared to the water and wood-chip samples (
Figure S9a). The organisms affiliated with
Plesiomonas and
Tolumonas were found in the roots of
S. tabernaemontani (13.3% and 10.0%, respectively), whereas
Azospira (13.8%) was found abundant in the
S. pectinata roots. The
T. angustifolia roots had organisms related to
Mycoplasma (2.3%),
Thauera (2.3%) and
Azospirillum (2.1%), while all of the genera in the roots of
D. spicata were less than 2%.
Azospira restricta (5.6%) and
Tolumonas auensis (9.9%) were the dominant species in the
T. angustifolia and
D. spicata roots, respectively (
Figure S9b).
In comparison with the media samples, the heatmap for the root samples demonstrated more individual groups, as
S. pectinata and
D. spicata were shown to be more similar to one another, while
S. tabernaemontani was different from the other three plants (
Figure S8).
Lu et al. [
5] have reviewed the microbial ecology of denitrification in wastewater systems and have discussed the common denitrifier genera that are found in denitrifying reactors. Based on the information provided by the authors, we selected the following genera as those potentially responsible for denitrification in our reactors and calculated the sum of the individual ratios from their taxa information. These selected populations included
Hyphomicrobium,
Paracoccus,
Azoarcus,
Thauera,
Methylophaga,
Pseudomonas,
Acidovorax,
Methyloversatilis,
Thiobacillus and
Comamonas.
The fold-changes of the expressed
nirS gene and nitrate removal efficiencies are presented in
Figure 8a [
2], while the sum of the denitrifier genera found in the water samples from the
T. angustifolia reactor and their change during the operation time are shown in
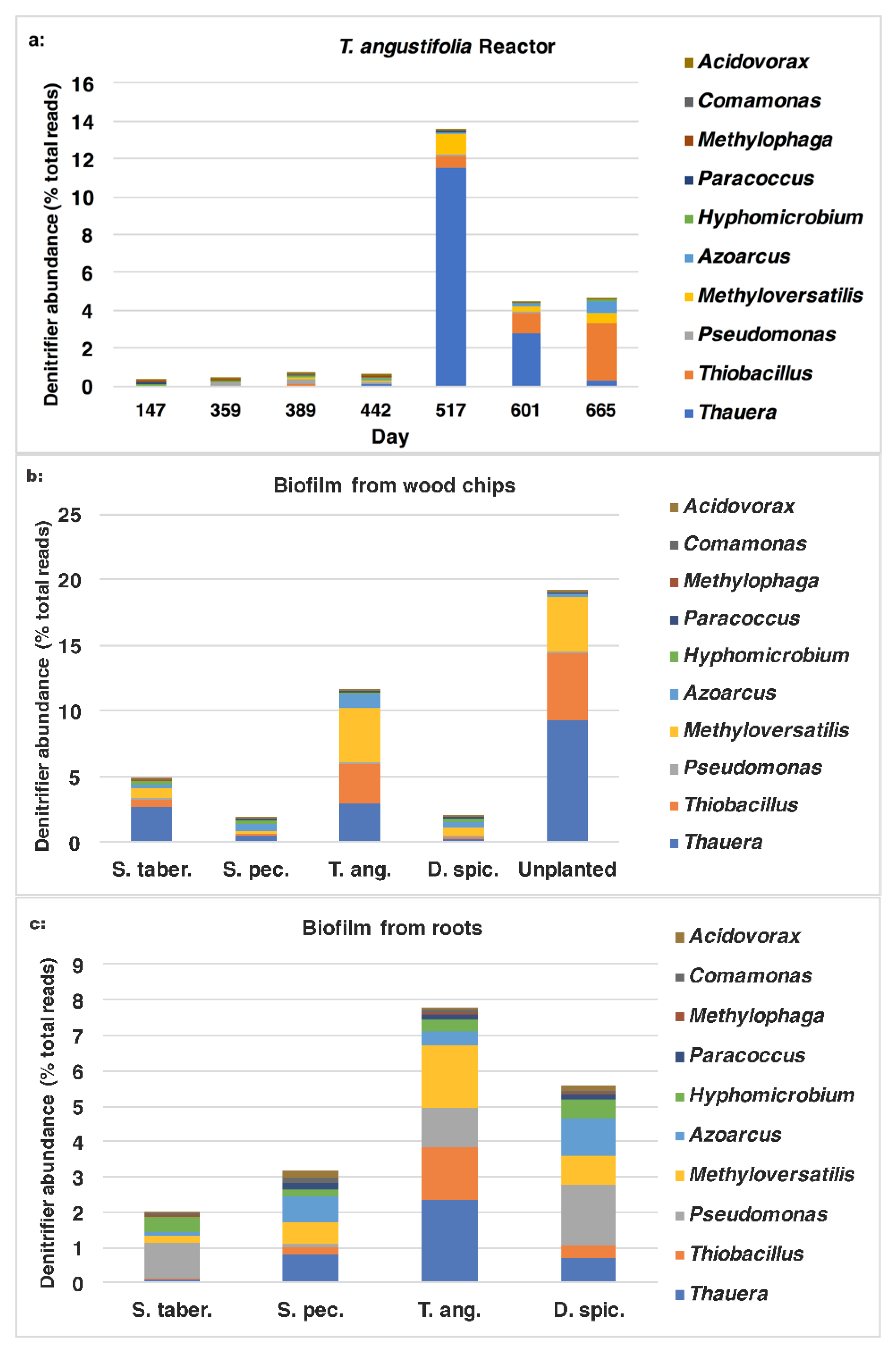
Figure 8b. These figures demonstrate the positive correlation between the higher denitrifier abundance and the greater
nirS populations on day 517 and their gradual decrease to the end of the operation. In both figures, the water samples from the top depth demonstrated larger populations of denitrifiers and those expressing the
nirS gene on days 517 and 601.
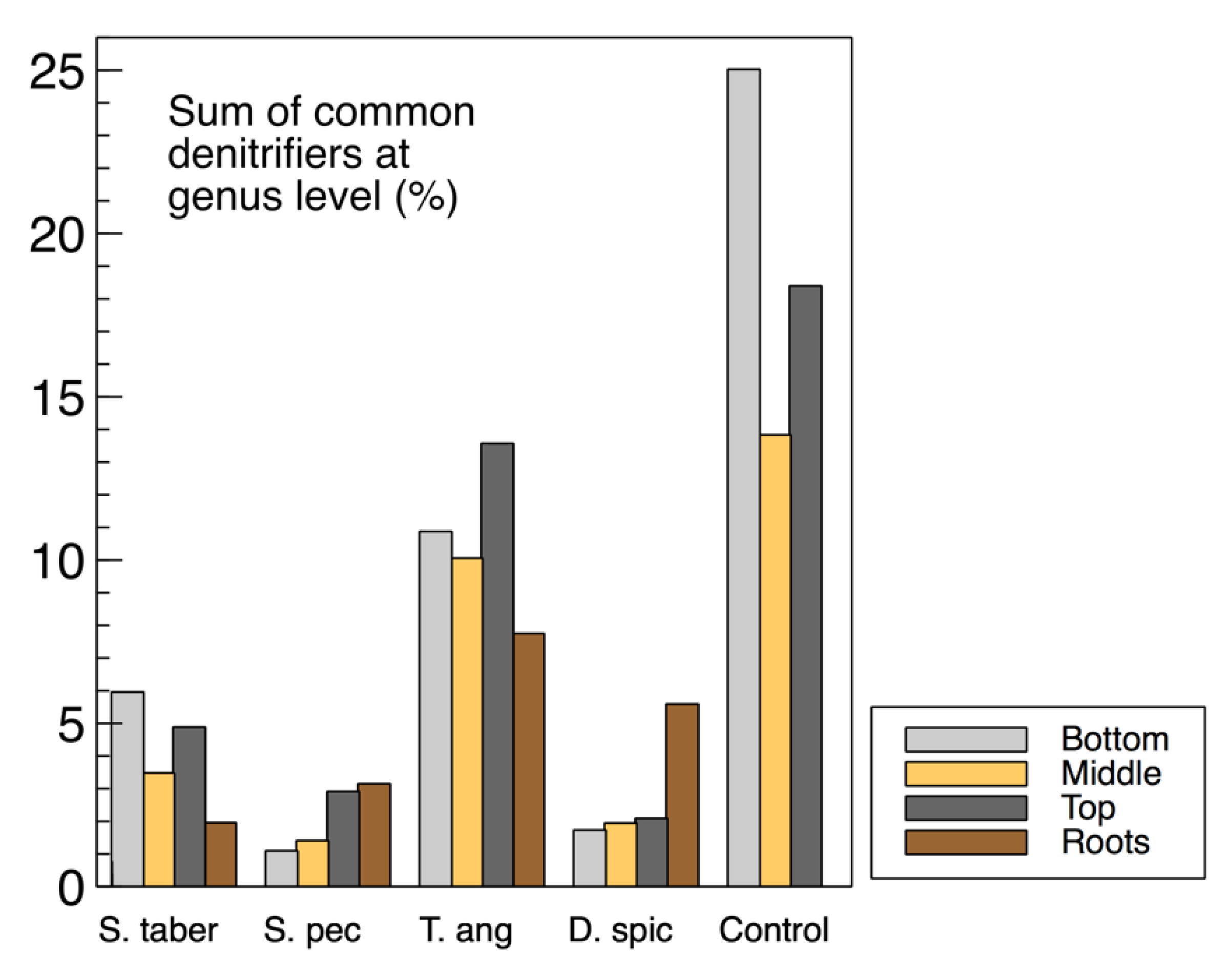
The sum of common denitrifiers in the biofilm samples from the media (wood chips) and plant roots are displayed in
Figure 9, while the
nirS,
nosZ and 16S rRNA genes were discussed earlier (
Figure 3). Some similarities were observed between the biofilm denitrifier populations and those expressing the
nosZ gene. In both methods, the
S. tabernaemontani and
T. angustifolia roots contained the smallest and largest denitrifier populations among the root samples, respectively. Moreover, the media samples from the control reactor showed the greatest abundance, followed by the
T. angustifolia unit. As well, the patterns of the two bioreactors for
nosZ copies through the reactor depth were similar to the denitrifier abundance, showing the greatest copies and abundance at the top depth in the
T. angustifolia reactor and at the bottom depth for the unplanted unit.
As mentioned earlier, the higher denitrifier ratio in the
T. angustifolia bioreactor can be attributed to the contributions of the plant’s rhizosphere and the root exudates. Nonetheless, unlike the mixed microbial activity results for the wood-chip media (
Figure 1a), a consistent trend of denitrifier abundance through reactor depth was not found in all the bioreactors. This finding indicates that, although the rhizosphere of
T. angustifolia likely had an important effect on the higher
nosZ copies and denitrifier population at the shallowest depth, the other plants did not provide a similar effect in their respective reactors. Furthermore, as the consistently greater activities that were observed at the top depth in these reactors did not correlate with the abundance of the classified organisms via 16S amplicon sequencing, these higher mixed microbial activities may have been caused by other microbial communities in these open reactors.
In addition to the sum quantity of the denitrifier populations comprised of the 10 genera mentioned earlier, it is of interest to investigate which microorganisms were found in greater proportions and had a larger contribution to the overall performance of the bioreactors. The ratios of each individual genus are demonstrated in
Figure 10a–c. In the water samples from the
T. angustifolia reactor (
Figure 10a) on day 517, the major organisms belonged to
Thauera (11.6%) and, to a lesser extent,
Methyloversatilis (1.1%),
Thiobacillus (0.6%) and
Azoarcus (0.1%). As the operation progressed, the communities affiliated with
Thauera and
Methyloversatilis decreased to 0.3% and 0.5%, respectively, while
Thiobacillus and
Azoarcus increased to 3.0% and 0.7%, respectively.
For the wood-chip samples (
Figure 10b), the majority of the organisms in the two bioreactors with the highest nitrate-reduction performances were affiliated with
Thauera,
Thiobacillus and
Methyloversatilis and, to a smaller degree,
Azoarcus. The abundance of
Thauera and
Thiobacillus were higher for the unplanted reactor’s media compared to the
T. angustifolia unit (9.3% vs. 3.0% and 5.1% vs. 3.0% respectively).
Similarly,
Thauera (2.3%),
Thiobacillus (1.8%) and
Thiobacillus (1.5%) were the major genera in the biofilm detached from the
T. angustifolia roots (
Figure 10c). The populations related to
Pseudomonas were another member of the denitrifiers that were found as the dominant denitrifier genus in the
S. tabernaemontani (1.0%) and
D. spicata (1.7%) roots.
These results demonstrate the importance of the populations associated with the genera
Thauera,
Thiobacillus and
Methyloversatilis, as these were the predominant denitrifying communities. Moreover,
Sulfurimonas,
Sulfurospirillum,
Methylotenera,
Bacillus and
Geobacter were the other general abundant genera that formed the community structure of the reactors with high denitrification performance.
Thiobacillus and
Sulfurimonas were the most commonly reported sulfur-oxidizing autotrophic denitrifiers, and
Methyloversatilis,
Thauera,
Thiobacillus and
Geobacter were found as the dominant genera in systems where methanol, ethanol or sulfur were the electron donors in the denitrification process [
42]. These findings suggest that both autotrophic denitrification pathways through organisms related to
Thiobacillus and those belonging to
Sulfurimonas (based on sulfur oxidation), as well as heterotrophic denitrification facilitated by
Thauera and methylotrophs such as
Methyloversatilis spp., may have played significant roles in our denitrifying bioreactors.
Moreover,
Sulfurospirillum spp. are known to be versatile and are able to grow organotrophically and chemolithotrophically in environments where organic and inorganic electron donors are present [
32]. The presence of organisms related to
Sulfurospirillum, which are known for their ability to reduce sulfur [
43,
44], as well as sulfate-reducing bacteria such as
Desulfotomaculum spp. [
35] and species affiliated with
Sulfurimonas with sulfur- and thiosulfate-oxidizing capacity [
34], indicate the development of a rich and complex sulfur cycle in our bioreactors.
Furthermore, the methylotrophic populations related to
Methyloversatilis and
Hyphomicrobium spp. were reported in denitrifying reactors metabolizing methanol [
45]. Methylotrophic bacteria are capable of utilizing methanol or methane as their source of energy and, therefore, can potentially reduce the level of the greenhouse gas emissions [
46]. The production of methane in wood-based denitrifying bioreactors has been previously reported (e.g., [
29,
47]). The microbial community sequencing results in this study indicate that, although methane may have been produced via the breakdown of the organic carbon in the wood biomass [
48], the types of the abundant bacteria developed in the reactors were capable of utilizing some of the produced methane as a source of energy for nitrate reduction.
This study demonstrated the superior role of
T. angustifolia in comparison with the other plant species to enhance the biological processes and facilitate denitrification in a shorter period of time. However, it was noted that the unplanted reactor was also capable of hosting a diverse population of microorganisms, including common denitrifiers, many of which were similar to those found in the bacterial composition of the
T. angustifolia reactor. Therefore, the presence of populations with the capability to decompose wood products (such as
Geobacillus spp.) may be an equally important factor in successful denitrification in wood-based systems. The fungi species are also known to contribute to denitrification directly [
49] or by degrading wood compounds and facilitating bacterial denitrification [
50,
51]. The interaction between the wood-decaying fungi and the bacteria found in wood is, however, poorly understood [
52]. As such, it is recommended to further investigate the presence and function of the fungi species in wood-chip bioreactors in order to better understand how these two communities originating from different phyla may aid denitrification by causing wood degradation or other potential mechanisms.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}