Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
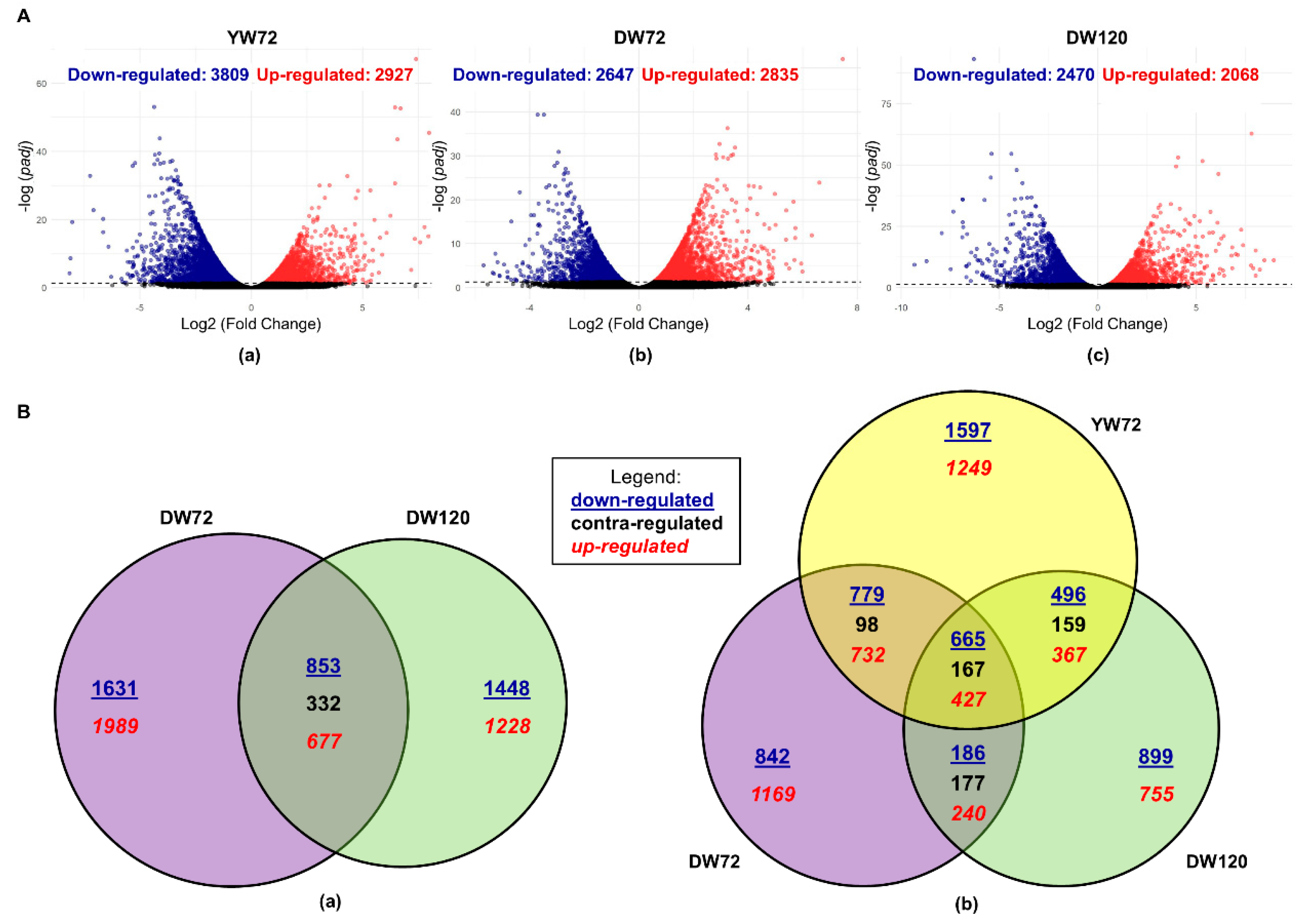
2.1. Differentially Expressed Genes of Moderately-Tolerant Barley at 72 h of Waterlogging
2.2. Differentially Expressed Genes of Tolerant Barley at 72 and 120 h of Waterlogging
2.3. Differentially Expressed Genes of Moderately-Tolerant and Tolerant Barleys under Waterlogging
2.4. Validation of Differentially Expressed Genes
2.5. Gene Ontology Term Enrichment for All Differentially Expressed Genes at 72 and 120 h of Waterlogging
2.6. Gene Ontology Term Enrichment of Down and Up-Regulated Genes at 72 and 120 h of Waterlogging
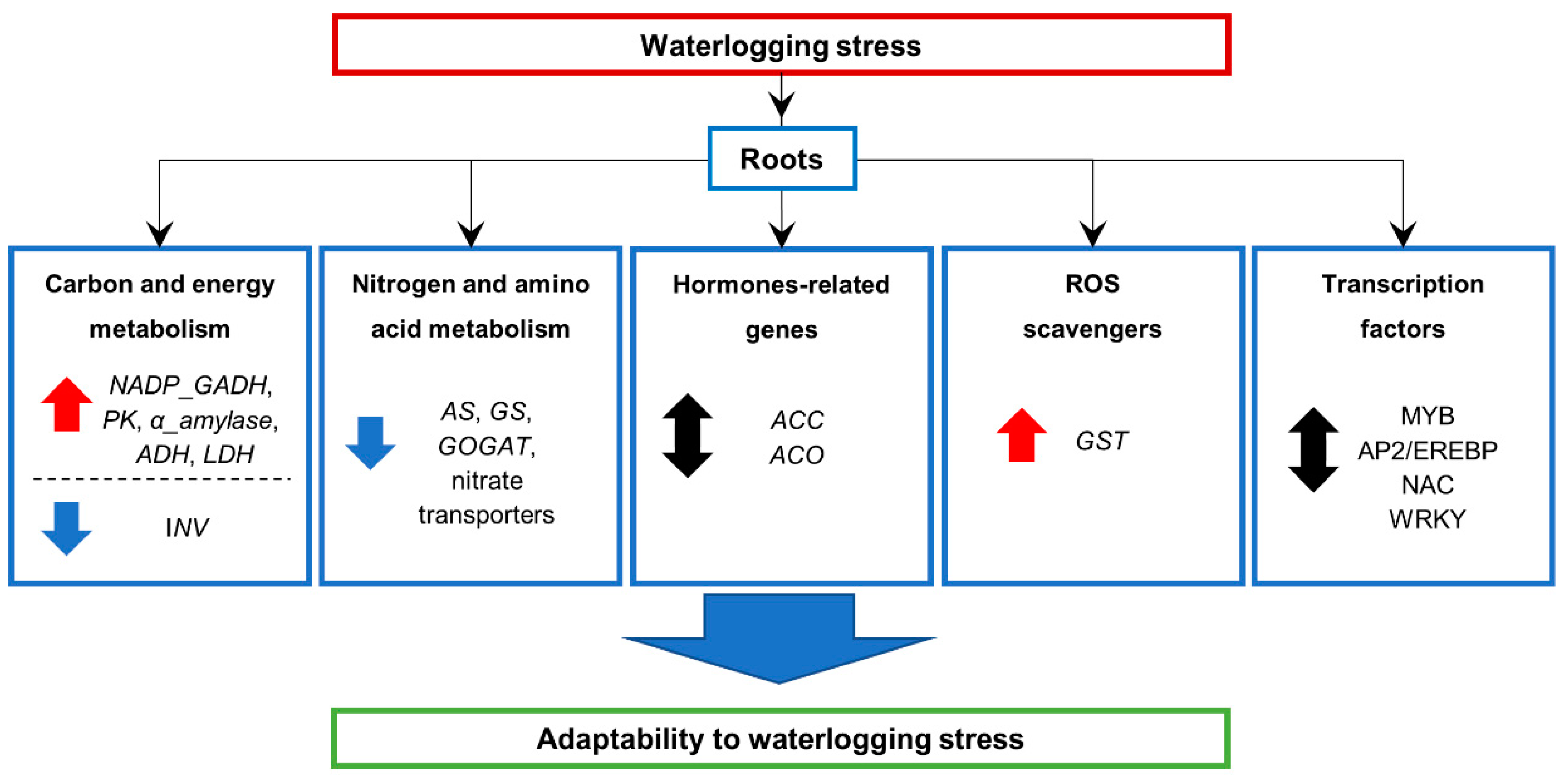
2.7. Expression of Genes Involved in Energy-Consuming Biosynthesis and Metabolism
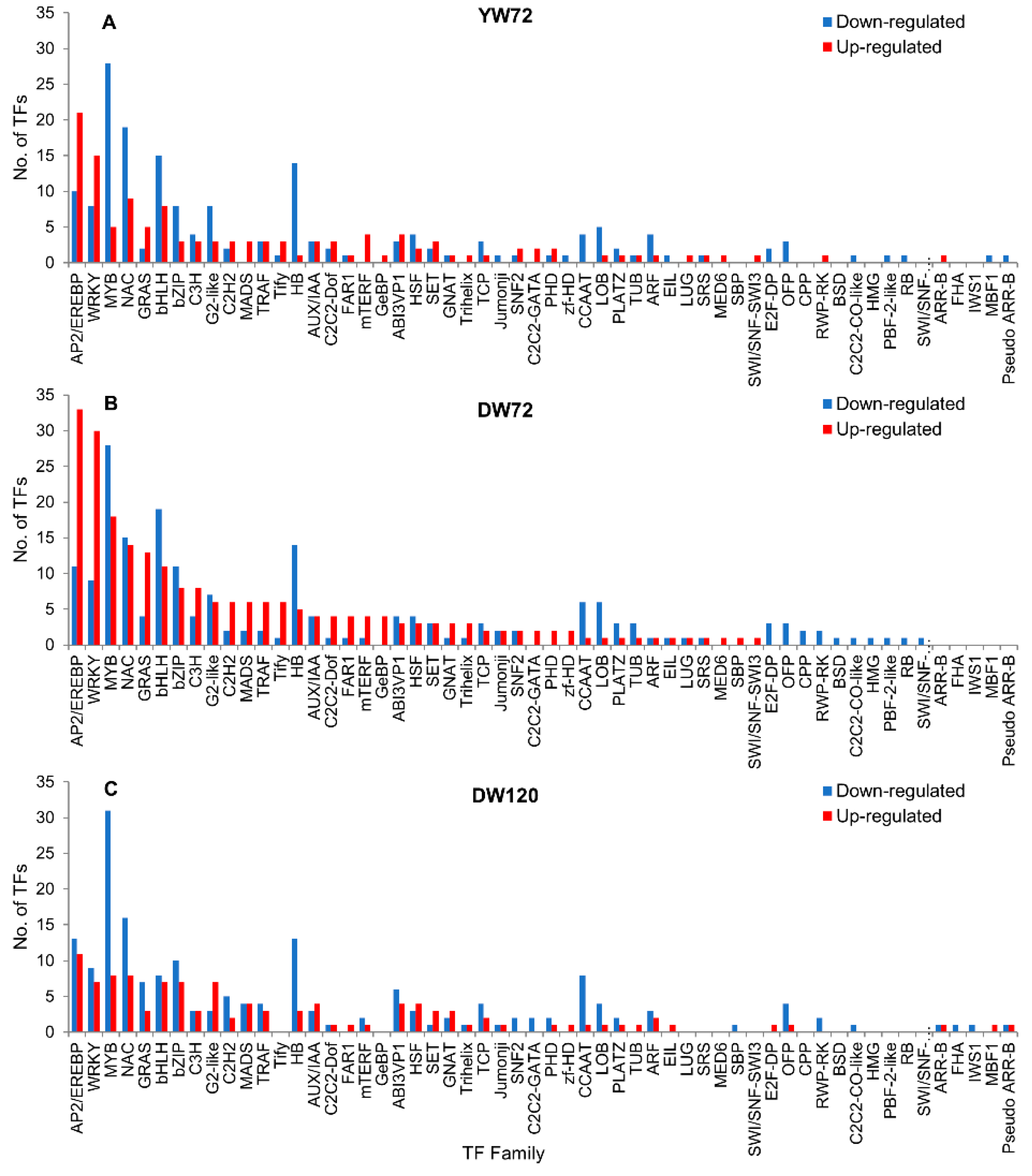
2.8. Hormone Related Genes and Transcription Factor Genes
3. Discussion
3.1. Global Gene Transcription Changes in Waterlogged Barley Root
3.2. Waterlogging Stress Up-Regulated Genes Related to Response to Abiotic Stresses
3.3. Effects on ROS Production
3.4. Effects on Energy-Consuming Biosynthetic Processes
3.5. Effects on Carbon Metabolism
3.6. Effects on Nitrogen and Amino Acid Metabolism
3.7. Transcription Factors Responses to Waterlogging
4. Materials and Methods
4.1. Plant Material and Waterlogging Treatment
4.2. RNA Extraction, Library Preparation, and Sequencing
4.3. Differential Expression Analysis
4.4. Quantitative Real-Time PCR (qRT-PCR) Validation
4.5. Pathway Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAOSTAT Production. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 18 November 2019).
- Statistics Canada. Estimated areas, yield, production, average farm price and total farm value of principal field crops. Available online: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=3210035901 (accessed on 18 November 2019).
- Canadian Agri-Food Trade Alliance (CAFTA). Available online: http://cafta.org/agri-food-exports/cafta-exports/ (accessed on 18 November 2019).
- Setter, T.L.; Waters, I. Review of prospects for germplasm improvement for waterlogging tolerance in wheat, barley and oats. Plant Soil 2003, 253, 1–34. [Google Scholar] [CrossRef]
- Pang, J.; Zhou, M.; Mendham, N.; Shabala, S. Growth and physiological responses of six barley genotypes to waterlogging and subsequent recovery. Aust. J. Agric. Res. 2004, 55, 895–906. [Google Scholar] [CrossRef]
- Malik, A.I.; Colmer, T.D.; Lambers, H.; Schortemeyer, M. Changes in physiological and morphological traits of roots and shoots of wheat in response to different depths of waterlogging. Aust. J. Plant Physiol. 2001, 28, 1121–1131. [Google Scholar] [CrossRef]
- Drew, M.C.; Sisworo, E.J. Early effects of flooding on nitrogen deficiency and leaf chlorosis in barley. New Phytol. 1977, 79, 567–571. [Google Scholar] [CrossRef]
- Setter, T.L.; Burgess, P.; Waters, I.; Kuo, J. Genetic diversity of barley and wheat for waterlogging tolerance in Western Australia. In Proceedings of the 9th Australian Barley Technical Symposium, Melbourne, VIC, Australia, 12–16 September 1999. [Google Scholar]
- Xiao, Y.; Wei, K.; Chen, J.; Zhou, M.; Zhang, G. Genotypic difference in growth inhibition and yield loss in barley under waterlogging stress. J. Zhejiang Univ. Sci. (Agric. Life Sci.) 2007, 33, 525–532. [Google Scholar]
- San Celedonio, R.P.; Abeledo, L.G.; Miralles, D.J. Identifying the critical period for waterlogging on yield and its components in wheat and barley. Plant Soil 2014, 378, 265–277. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Fukao, T.; Gibbs, D.J.; Holdsworth, M.J.; Lee, S.C.; Licausi, F.; Perata, P.; Voesenek, L.A.; van Dongen, J.T. Making sense of low oxygen sensing. Trends Plant Sci. 2012, 17, 129–138. [Google Scholar] [CrossRef]
- Borrego-Benjumea, A.; Carter, A.; Glenn, A.J.; Badea, A. Impact of excess moisture due to precipitation on barley grain yield in the Canadian Prairies. Can. J. Plant Sci. 2019, 99, 93–96. [Google Scholar] [CrossRef]
- Malik, A.I.; Colmer, T.D.; Lambers, H.; Setter, T.L.; Schortemeyer, M. Short-term waterlogging has long-term effects on the growth and physiology of wheat. New Phytol. 2002, 153, 225–236. [Google Scholar] [CrossRef]
- Ahmed, F.; Rafii, M.; Ismail, M.R.; Juraimi, A.S.; Rahim, H.; Asfaliza, R.; Latif, M. Waterlogging tolerance of crops: Breeding, mechanism of tolerance, molecular approaches, and future prospects. Biomed. Res. Int. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A. Improving Flooding Tolerance of Crop Plants. Agronomy 2018, 8, 160. [Google Scholar] [CrossRef]
- Parent, C.; Capelli, N.; Berger, A.; Crèvecœur, M.; Dat, J.F. An overview of plant responses to soil waterlogging. Plant Stress 2008, 2, 20–27. [Google Scholar]
- van Dongen, J.T.; Licausi, F. Oxygen sensing and signaling. Annu. Rev. Plant Biol. 2015, 66, 345–367. [Google Scholar] [CrossRef]
- Kumutha, D.; Sairam, R.K.; Ezhilmathi, K.; Chinnusamy, V.; Meena, R.C. Effect of waterlogging on carbohydrate metabolism in pigeon pea (Cajanus cajan L.): Upregulation of sucrose synthase and alcohol dehydrogenase. Plant Sci. 2008, 175, 706–716. [Google Scholar] [CrossRef]
- Dennis, E.S.; Dolferus, R.; Ellis, M.; Rahman, M.; Wu, Y.; Hoeren, F.U.; Grover, A.; Ismond, K.P.; Good, A.G.; Peacock, W.J. Molecular strategies for improving waterlogging tolerance in plants. J. Exp. Bot. 2010, 51, 89–97. [Google Scholar] [CrossRef]
- Kuai, J.; Chen, Y.; Wang, Y.; Meng, Y.; Chen, B.; Zhao, W.; Zhou, Z. Effect of waterlogging on carbohydrate metabolism and the quality of fiber in cotton (Gossypium hirsutum L.). Front. Plant Sci. 2016, 7, 877. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, V.; Pawar, S.K.; Singh, P.K.; Kaur, A.; Sharma, D. Abiotic stress and wheat grain quality: A comprehensive review. In Wheat Production in Changing Environments; Hasanuzzaman, M., Nahar, K., Hossain, M., Eds.; Springer: Singapore, 2019; pp. 63–87. [Google Scholar] [CrossRef]
- Xu, Q.T.; Yang, L.; Zhou, Z.Q.; Mei, F.Z.; Qu, L.H.; Zhou, G.S. Process of aerenchyma formation and reactive oxygen species induced by waterlogging in wheat seminal roots. Planta 2013, 238, 969. [Google Scholar] [CrossRef] [PubMed]
- Shiono, K.; Ejiri, M.; Shimizu, K.; Yamada, S. Improved waterlogging tolerance of barley (Hordeum vulgare) by pretreatment with ethephon. Plant Prod. Sci. 2019, 22, 285–295. [Google Scholar] [CrossRef]
- Komatsu, S.; Shirasaka, N.; Sakata, K. ‘Omics’ techniques for identifying flooding-response mechanisms in soybean. J. Proteome Res. 2013, 93, 169–178. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, J.; He, X.; Sun, H.; Zhang, G.; Wu, F. Comparative proteomic analysis of drought tolerance in the two contrasting Tibetan wild genotypes and cultivated genotype. BMC Genom. 2015, 16, 432–451. [Google Scholar] [CrossRef]
- Meloni, D.A.; Oliva, M.O.; Martinez, C.A.; Cambraia, J. Photosynthesis and activity of superoxide dismutase, peroxidase and glutathione reductase in cotton under salt stress. Environ. Exp. Bot. 2003, 49, 69–76. [Google Scholar] [CrossRef]
- Dixon, D.P.; Edwards, R. Glutathione Transferases. Arabidopsis Book 2010, 8, e0131. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liang, K.; Fang, T.; Zhao, H.; Han, X.; Cai, M.; Qiu, F. A group VII ethylene response factor gene, ZmEREB180, coordinates waterlogging tolerance in maize seedlings. Plant Biotechnol. J. 2019, 17, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Vaillancourt, R.; Mendham, N.; Zhou, M. Comparative mapping of quantitative trait loci associated with waterlogging tolerance in barley (Hordeum vulgare L.). BMC Genom. 2008, 9, 401. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, G.; Shabala, S.; Koutoulis, A.; Shabala, L.; Johnson, P.; Li, C.; Zhou, M. Identification of aerenchyma formation related QTL in barley that can be effective in breeding for waterlogging tolerance. Theor. Appl. Genet. 2016, 129, 1167–1177. [Google Scholar] [CrossRef]
- Gill, M.B.; Zeng, F.; Shabala, L.; Zhang, G.; Fan, Y.; Shabala, S.; Zhou, M. Cell-based phenotyping reveals QTL for membrane potential maintenance associated with hypoxia and salinity stress tolerance in barley. Front. Plant. Sci. 2017, 8, 1941. [Google Scholar] [CrossRef]
- Broughton, S.; Zhou, G.; Teakle, N.; Matsuda, R.; Zhou, M.; O’Leary, R.; Colmer, T.; Li, C. Waterlogging tolerance is associated with root porosity in barley (Hordeum vulgare L.). Mol. Breed. 2015, 35, 1–15. [Google Scholar] [CrossRef]
- Gill, M.B.; Zeng, F.; Shabala, L.; Zhang, G.; Yu, M.; Demidchik, V.; Shabala, S.; Zhou, M. Identification of QTL related to ROS formation under hypoxia and their association with waterlogging and salt tolerance in barley. Int. J. Mol. Sci. 2019, 20, 699. [Google Scholar] [CrossRef]
- Zhang, X.; Shabala, S.; Koutoulis, A.; Shabala, L.; Zhou, M. Meta-analysis of major QTL for abiotic stress tolerance in barley and implications for barley breeding. Planta 2017, 245, 283–295. [Google Scholar] [CrossRef]
- Lasanthi-Kudahettige, R.; Magneschi, L.; Loreti, E.; Gonzali, S.; Licausi, F.; Novi, G.; Beretta, O.; Vitulli, F.; Alpi, A.; Perata, P. Transcript profiling of the anoxic rice coleoptile. Plant Physiol. 2007, 144, 218–231. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, L.; Zou, X.; Tao, Y.; Liu, Z.; Zheng, Y. Submergence responsive microRNAs are potentially involved in the regulation of morphological and metabolic adaptations in maize root cells. Ann. Bot. 2008, 102, 509–519. [Google Scholar] [CrossRef]
- Kreuzwieser, J.; Hauberg, J.; Howell, K.A.; Carroll, A.; Rennenberg, H.; Millar, A.H.; Whelan, J. Differential response of gray poplar leaves and roots underpins stress adaptation during hypoxia. Plant Physiol. 2009, 149, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S.; Wilson, I.W. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.). Plant Cell Physiol. 2010, 51, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, Y.; Maruyama, K.; Yasue, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Komatsu, S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol. 2011, 77, 129–144. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Qi, X.; Li, D.; Wei, W.; Zhang, X. Global gene expression responses to waterlogging in roots of sesame (Sesamum indicum L.). Acta Physiol. Plant 2012, 34, 2241–2249. [Google Scholar] [CrossRef]
- Xu, X.; Chen, M.; Ji, J.; Xu, Q.; Qi, X.; Chen, X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017, 17, 129. [Google Scholar] [CrossRef]
- Najeeb, U.; Bange, M.P.; Tan, D.K.Y.; Atwell, B.J. Consequences of waterlogging in cotton and opportunities for mitigation of yield losses. AoB Plants 2015, 7, plv080. [Google Scholar] [CrossRef]
- Luan, H.; Guo, B.; Pan, Y.; Lv, C.; Shen, H.; Xu, R. Morpho-anatomical and physiological responses to waterlogging stress in different barley (Hordeum vulgare L.) genotypes. Plant Growth Regul. 2018, 85, 399–409. [Google Scholar] [CrossRef]
- Luan, H.; Shen, H.; Pan, Y.; Guo, B.; Lv, C.; Xu, R. Elucidating the hypoxic stress response in barley (Hordeum vulgare L.) during waterlogging: A proteomics approach. Sci. Rep. 2018, 8, 9655. [Google Scholar] [CrossRef]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; Thirunavukkarasu, N. RNAseq revealed the important gene pathways controlling adaptive mechanisms under waterlogged stress in maize. Sci. Rep. 2017, 7, 10950. [Google Scholar] [CrossRef]
- Hsu, S.-K.; Tung, C.-W. RNA-Seq analysis of diverse rice genotypes to identify the genes controlling coleoptile growth during submerged germination. Front. Plant Sci. 2017, 8, 762. [Google Scholar] [CrossRef] [PubMed]
- Nafisi, M.; Stranne, M.; Fimognari, L.; Atwell, S.; Martens, H.J.; Pedas, P.R.; Hansen, S.F.; Nawrath, C.; Scheller, H.V.; Kliebenstein, D.J.; et al. Acetylation of cell wall is required for structural integrity of the leaf surface and exerts a global impact on plant stress responses. Front. Plant Sci. 2015, 6, 550. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Aravind, L. Novel eukaryotic enzymes modifying cell-surface biopolymers. Biol. Direct. 2010, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, L.; Zhai, H.; Song, X.; He, S.; Liu, Q. A novel α/β-hydrolase gene IbMas enhances salt tolerance in transgenic sweetpotato. PLoS ONE 2014, 9, e115128. [Google Scholar] [CrossRef]
- Oh, M.W.; Komatsu, S. Characterization of proteins in soybean roots under flooding and drought stresses. J. Proteom. 2015, 114, 161–181. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Y.; Li, Z.; Shi, W.; Gao, F.; Zhou, Y.; Zhang, G.; Feng, J. Genome-Wide Identification and Expression Analyses of the Chitinases under Cold and Osmotic stress in Ammopiptanthus nanus. Genes 2019, 10, 472. [Google Scholar] [CrossRef]
- Liu, H.Z.; Wang, X.E.; Zhang, H.J.; Yang, Y.Y.; Ge, X.C.; Song, F.M. A rice serine carboxypeptidase-like gene OsBISCPL1 is involved in regulation of defense responses against biotic and oxidative stress. Gene 2008, 420, 57–65. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, H.; Pan, Y.; Yu, Y.; Luan, S.; Li, L. A DTX/MATE-type transporter facilitates abscisic acid efflux and modulates ABA sensitivity and drought tolerance in Arabidopsis. Mol. Plant 2014, 7, 1522–1532. [Google Scholar] [CrossRef]
- Ogawa, I.; Nakanishi, H.; Mori, S.; Nishizawa, N.K. Time course analysis of gene regulation under cadmium stress in rice. Plant Soil 2009, 325, 97. [Google Scholar] [CrossRef]
- Ren, H.; Gray, W.M. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, Q.; Hu, Z.; Sun, X.; Fan, S.; Zhang, H. Function of the auxin-responsive gene TaSAUR75 under salt and drought stress. Crop J. 2018, 6, 181–190. [Google Scholar] [CrossRef]
- Singh, P.K.; Indoliya, Y.; Chauhan, A.S.; Singh, S.P.; Singh, A.P.; Dwivedi, S.; Tripathi, R.D.; Chakrabarty, D. Nitric oxide mediated transcriptional modulation enhances plant adaptive responses to arsenic stress. Sci Rep. 2017, 7, 3592. [Google Scholar] [CrossRef] [PubMed]
- Wink, M. Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochem. 2003, 64, 3–19. [Google Scholar] [CrossRef]
- Liu, H.; Du, Y.; Li, H.; Wang, X.Y.; Yu, Z.Y.; Zhu, W.N.; Zhang, D.P.; Zhang, L.S. Cloning, expression and functional analysis of WDHN1 gene from wheat (Triticum aestivum). J. Agric. Biotech. 2016, 24, 1676–1687. [Google Scholar]
- Blokhina, O.; Virolainen, E.; Fagerstedt, K.V. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Liu, J.; Dai, T.; Jing, Q.; Cao, W.; Jiang, D. Alterations in photosynthesis and antioxidant enzyme activity in winter wheat subjected to post-anthesis water-logging. Photosynthetica 2008, 46, 21–27. [Google Scholar] [CrossRef]
- Qi, X.; Li, Q.; Ma, X.; Qian, C.; Wang, H.; Ren, N.; Shen, C.; Huang, S.; Xu, X.; Xu, Q.; et al. Waterlogging-induced adventitious root formation in cucumber is regulated by ethylene and auxin through reactive oxygen species signalling. Plant Cell. Environ. 2019, 42, 1458–1470. [Google Scholar] [CrossRef]
- Zhang, G.; Tanakamaru, K.; Abe, J.; Morita, S. Influence of waterlogging on some antioxidative enzymatic activities of two barley genotypes differing in anoxia tolerance. Acta Physiol. Plant 2007, 29, 171–176. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Lin, H.W.; Chem, R.H.; Lo, H.F.; Li, F. Reduction susceptibility to waterlogging together with highlight stress is related to increases in superoxide dismutase and catalase activities in sweet potato. Plant Growth Regul. 1999, 27, 167–172. [Google Scholar] [CrossRef]
- Zhang, X.; Shabala, S.; Koutoulis, A.; Shabala, L.; Johnson, P.; Hayes, D.; Nichols, D.S.; Zhou, M. Waterlogging tolerance in barley is associated with faster aerenchyma formation in adventitious roots. Plant Soil 2015, 394, 355–372. [Google Scholar] [CrossRef]
- Yordanova, R.Y.; Christov, K.N.; Popova, L.P. Antioxidative enzymes in barley plants subjected to soil flooding. Environ. Exp. Bot. 2004, 51, 93–101. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, K.S.; Jang, Y.S.; Hwang, J.H.; Lee, D.H.; Choi, I.H. Global gene expression responses to waterlogging in leaves of rape seedlings. Plant Cell Rep. 2014, 33, 289. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.H.; Xu, X.W.; Lin, X.J.; Zhang, W.J.; Chen, X.H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics 2012, 99, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, X.; Jiu, S.; Zhang, K.; Wang, C.; Fang, J. Analysis of the regulation networks in grapevine reveals response to waterlogging stress and candidate gene-marker selection for damage severity. R. Soc. 2018, 5, 172253. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, C.C.; Sachs, M.M. Molecular and cellular adaptations of maize to flooding stress. Ann. Bot. 2003, 91, 11–127. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.; Bailey-Serres, J. Selective mRNA translation tailors low oxygen energetics. In Low-Oxygen Stress in Plants; van Dongen, J.T., Licausi, F., Eds.; Springer-Verlag: Vienna, Austria, 2014; Volume 21, pp. 95–115. [Google Scholar] [CrossRef]
- Springer, B.; Werr, W.; Starlinger, P.; Bennett, D.C.; Freeling, M. The shrunken gene on chromosome 9 of Zea mays L. expressed in various plant tissues and encode an anaerobic protein. Mol. Gen. Genet. 1986, 220, 461–468. [Google Scholar] [CrossRef]
- Guglielminetti, L.; Perata, P.; Alpi, A. Effect of anoxia on carbohydrate metabolism in rice seedlings. Plant Physiol. 1995, 108, 735–741. [Google Scholar] [CrossRef]
- Coutinho, I.D.; Mertz-Henning, L.M.; Döpp, S.A.; Nepomuceno, A.L.; Moraes, L.A.C.; Marcolino-Gomes, J.; Richter, C.; Schwalbe, H.; Colnago, L.A. Flooded soybean metabolomic analysis reveals important primary and secondary metabolites involved in the hypoxia stress response and tolerance. Environ. Exp. Bot. 2018, 153, 176–187. [Google Scholar] [CrossRef]
- Bieniawska, Z. Functional Analysis of the Sucrose Synthase Gene Family in Arabidopsis thaliana. Ph.D. Thesis, University of Potsdam, Potsdam, Germany, 6 March 2006. [Google Scholar]
- Ismond, K.P.; Dolferus, R.; De Pauw, M.; Dennis, E.S.; Good, A.G. Enhanced low oxygen survival in Arabidopsis through increased metabolic flux in the fermentative pathway. Plant Physiol. 2003, 132, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzi, G.; Weichert, A.; Schnell, M.; Ruiz, C.; Bossard, S.; Blösch, R.; Plaza-Wüthrich, S.; Chanyalew, S.; Assefa, K.; Tadele, Z. Waterlogging affects plant morphology and the expression of key genes in tef (Eragrostis tef). Plant Direct 2018, 2, e00056. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, X.; Dai, J.; Luo, Z.; Li, Z.; Lu, H. Global gene expression in cotton (Gossypium hirsutum L.) leaves to waterlogging stress. PLoS ONE 2017, 12, e0185075. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Dong, S.; Zhao, B.; Liu, P.; Zhang, J. Responses of Nitrogen Metabolism, Uptake and Translocation of Maize to Waterlogging at Different Growth Stages. Front Plant Sci. 2017, 8, 1216. [Google Scholar] [CrossRef] [PubMed]
- Limami, A.M.; Glevarec, G.; Ricoult, C.; Cliquet, J.-B.; Planchet, E. Concerted modulation of alanine and glutamate metabolism in young Medicago truncatula seedlings under hypoxic stress. J. Exp. Bot. 2008, 59, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Pande, A.; Saxena, S.C.; Thapliyal, M.; Guru, S.K.; Kumar, A. Role of AP2/EREBP Transcription Factor Family in Environmental Stress Tolerance. Cell Cell. Lif. Sci. J. 2018, 3, 000120. [Google Scholar] [CrossRef]
- Nakashima, K.; Jan, A.; Todaka, D.; Maruyama, K.; Goto, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Comparative functional analysis of six drought-responsive promoters in transgenic rice. Planta 2014, 239, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar] [CrossRef]
- Shiono, K.; Yamauchi, T.; Yamazaki, S.; Mohanty, B.; Malik, A.I.; Nagamura, Y.; Nishizawa, N.; Tsutsumi, N.; Colmer, T.D.; Nakazono, M. Microarray analysis of laser-microdissected tissues indicates the biosynthesis of suberin in the outer part of roots during formation of a barrier to radial oxygen loss in rice (Oryza sativa). J. Exp. Bot. 2014, 65, 4795–4806. [Google Scholar] [CrossRef]
- Liu, Z.; Kumari, S.; Zhang, L.; Zheng, Y.; Ware, D. Characterization of miRNAs in response to short-term waterlogging in three inbred lines of Zea mays. PLoS ONE 2012, 7, e39786. [Google Scholar] [CrossRef]
- Yang, A.; Dai, X.; Zhang, W.-H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef]
- Zhao, N.; Li, C.; Yan, Y.; Cao, W.; Song, A.; Wang, H.; Chen, S.; Jiang, J.; Chen, F. Comparative transcriptome analysis of waterlogging-sensitive and waterlogging-tolerant Chrysanthemum morifolium cultivars under waterlogging stress and reoxygenation conditions. Int. J. Mol. Sci. 2018, 19, 1455. [Google Scholar] [CrossRef]
- Takeda, K. Varietal variation of flooding tolerance in barley seedlings, and its diallel analysis. Jpn. J. Breed. 1989, 39, 174–175. [Google Scholar]
- Daehwan, K.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chen, H.; Yi, T.; Daimon, C.M.; Boyle, J.P.; Peers, C.; Maudsley, S.; Martin, B. VennPlex—A novel Venn diagram program for comparing and visualizing datasets with differentially regulated datapoints. PLoS ONE 2013, 8, e53388. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler:A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, 191–198. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Gen. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]






© 2020 by Her Majesty the Queen in Right of Canada as represented by the Minister of Agriculture and Agri-Food Canada. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego-Benjumea, A.; Carter, A.; Tucker, J.R.; Yao, Z.; Xu, W.; Badea, A. Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.). Plants 2020, 9, 240. https://doi.org/10.3390/plants9020240
Borrego-Benjumea A, Carter A, Tucker JR, Yao Z, Xu W, Badea A. Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.). Plants. 2020; 9(2):240. https://doi.org/10.3390/plants9020240
Chicago/Turabian StyleBorrego-Benjumea, Ana, Adam Carter, James R. Tucker, Zhen Yao, Wayne Xu, and Ana Badea. 2020. "Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.)" Plants 9, no. 2: 240. https://doi.org/10.3390/plants9020240
APA StyleBorrego-Benjumea, A., Carter, A., Tucker, J. R., Yao, Z., Xu, W., & Badea, A. (2020). Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.). Plants, 9(2), 240. https://doi.org/10.3390/plants9020240