Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny

 , ,
, , 
Abstract
1. Introduction
2. Results
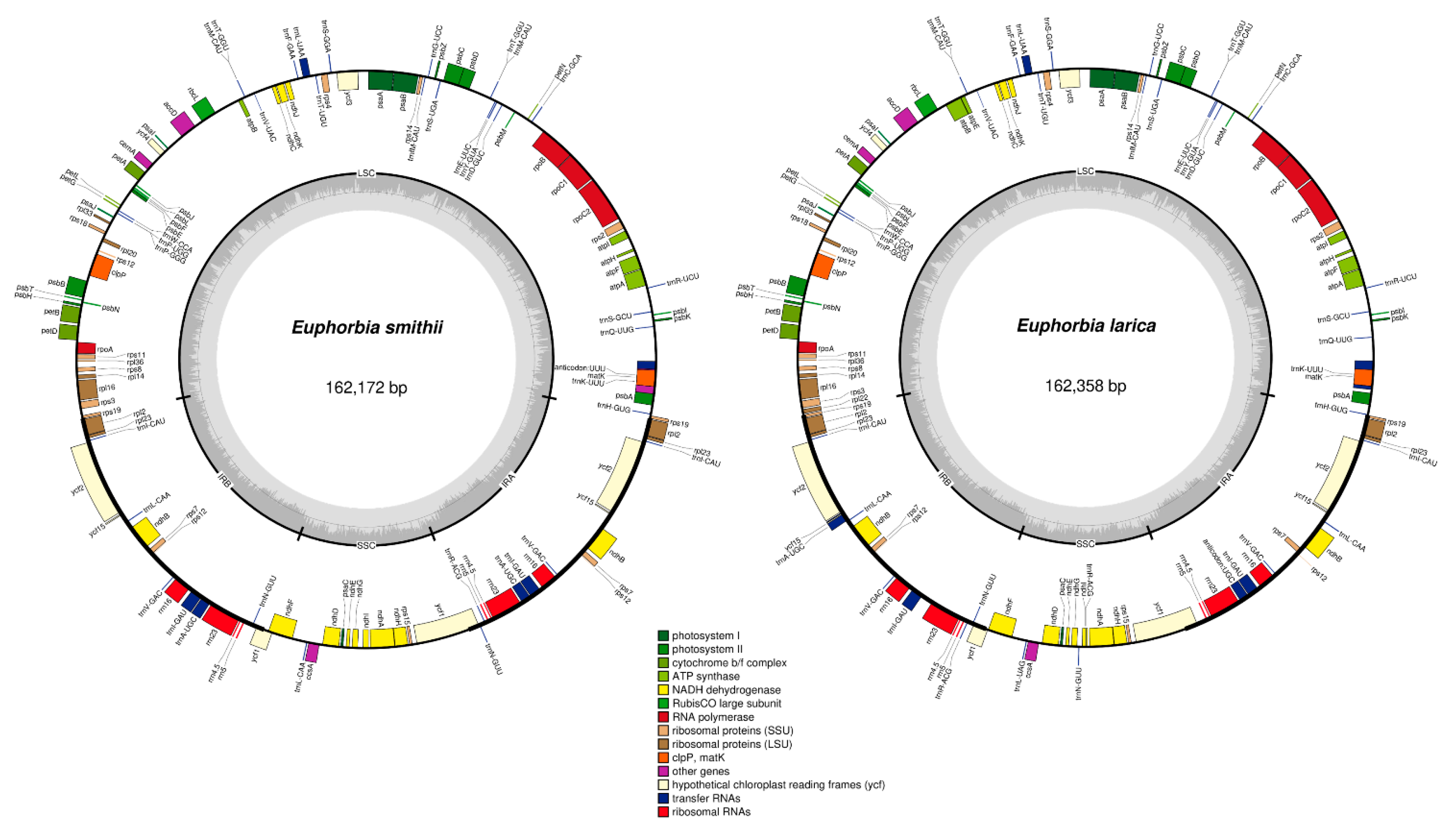
2.1. Comparative Characteristics of Chloroplast genomes
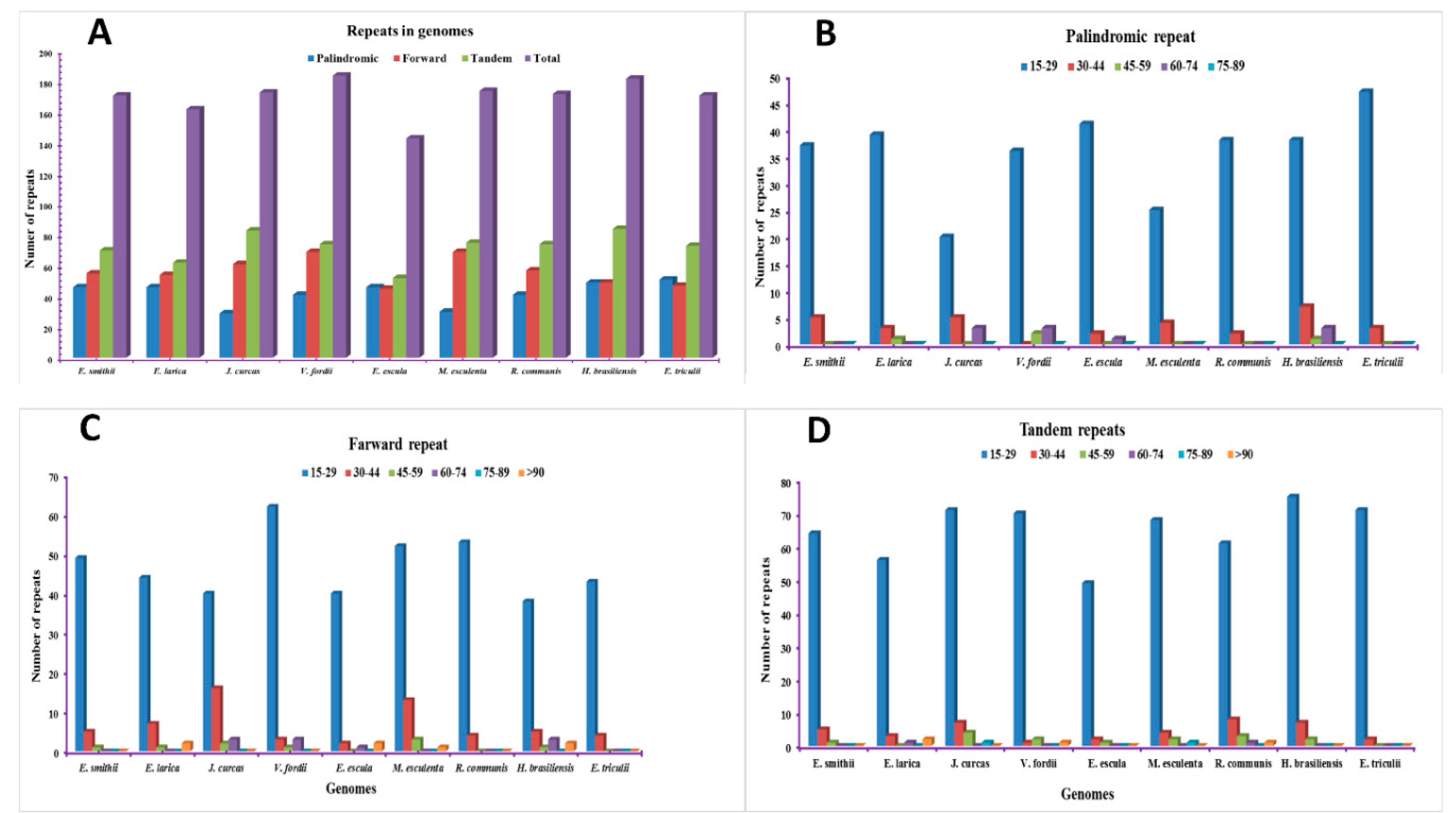
2.2. Analysis of Repetitive Sequences in the Genomes
2.3. SSRs Polymorphism Analysis
2.4. Compression and Augmentation of IR Region
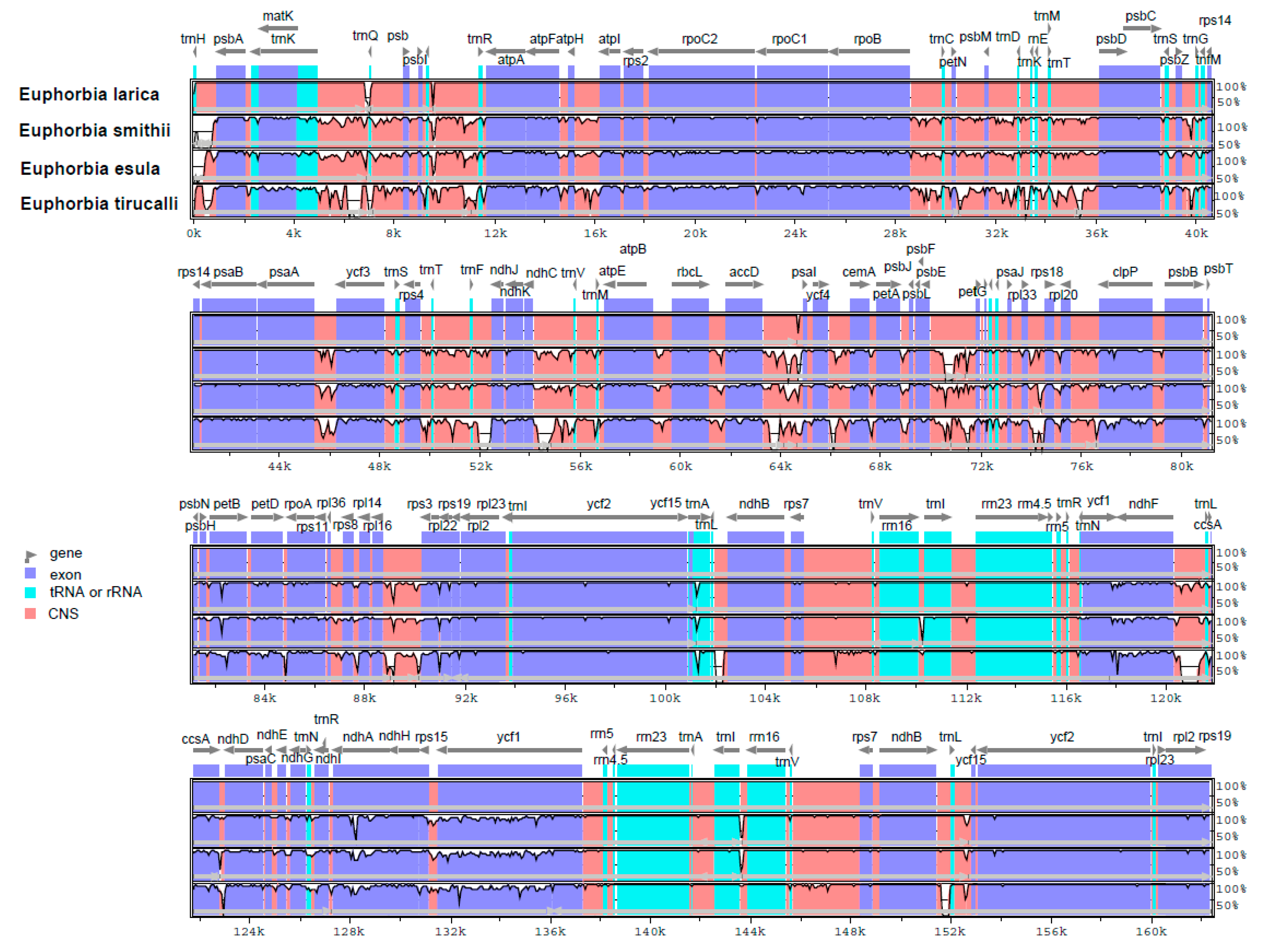
2.5. Comparison of the Hotspot Region in the cp Genome
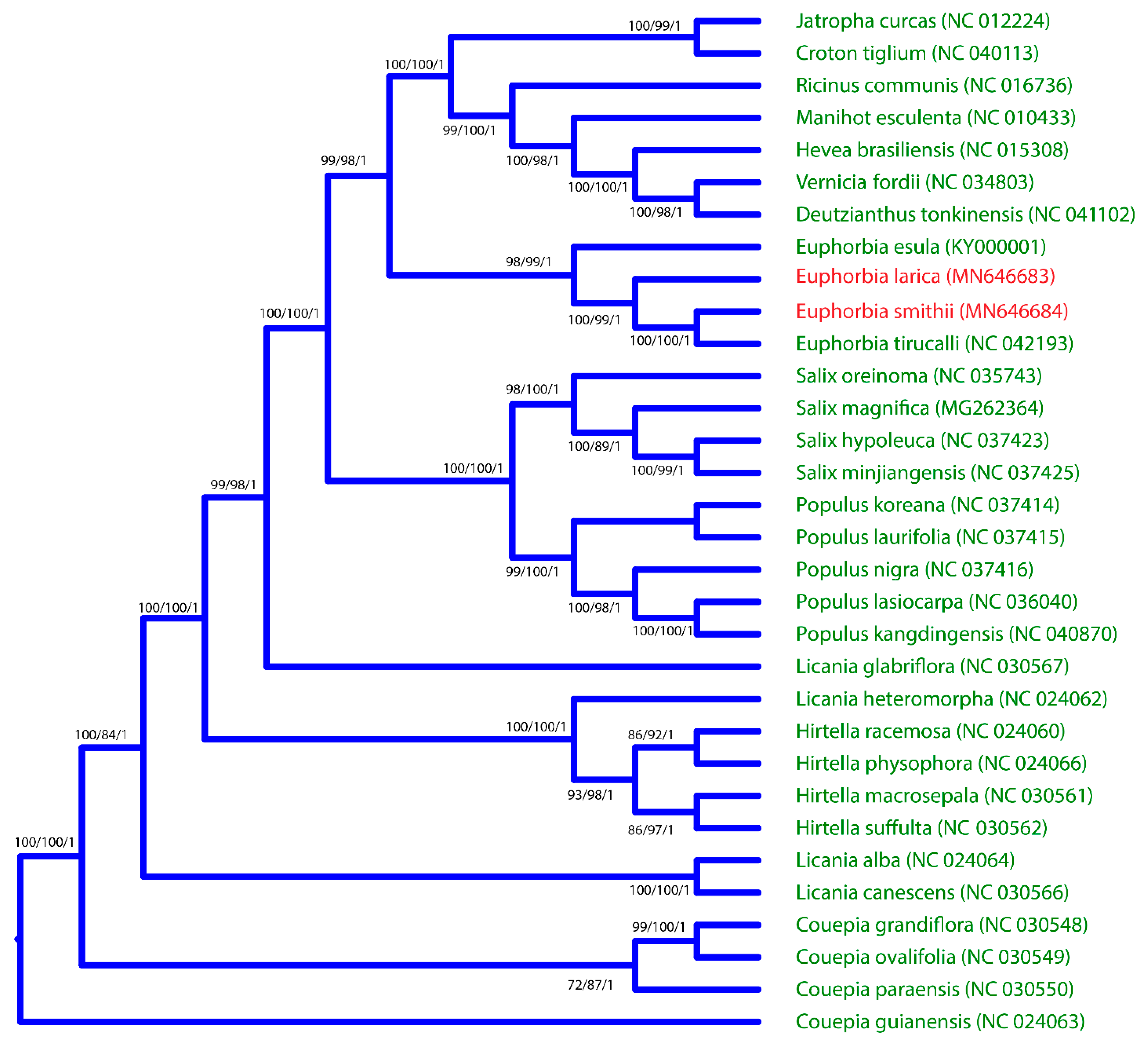
2.6. Phylogenomic Analysis of E. larica and E. smithii and Its Comparison with Related Species
3. Discussion
4. Material and Methods
4.1. Chloroplast DNA Extraction and Sequencing
4.2. Genome Assembly
4.3. Genome Annotation
4.4. Repeat Identification
4.5. Sequence Divergence and Phylogenetic Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bauer, J.; Chen, K.; Hiltbunner, A.; Wehrli, E.; Eugster, M.; Schnell, D.; Kessler, F. The major protein import receptor of plastids is essential for chloroplast biogenesis. Nature 2000, 403, 203. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast genome. Essays Biochem. 1995, 30, 49–57. [Google Scholar] [PubMed]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C. Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef] [PubMed]
- Dumolin, S.; Demesure, B.; Petit, R. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef]
- Schmidt, L.; Fischer, M.; Oja, T. Two closely related species differ in their regional genetic differentiation despite admixing. AoB Plants 2018, 10, ply007. [Google Scholar] [CrossRef]
- Horn, J.W.; van Ee, B.W.; Morawetz, J.J.; Riina, R.; Steinmann, V.W.; Berry, P.E.; Wurdack, K.J. Phylogenetics and the evolution of major structural characters in the giant genus Euphorbia, L.(Euphorbiaceae). Mol. Phylogenet. Evol. 2012, 63, 305–326. [Google Scholar] [CrossRef]
- Jassbi, A.R. Chemistry and biological activity of secondary metabolites in Euphorbia from Iran. Phytochemistry 2006, 67, 1977–1984. [Google Scholar] [CrossRef]
- Zimmermann, N.; Ritz, C.M.; Hellwig, F. Further support for the phylogenetic relationships within Euphorbia L. (Euphorbiaceae) from nrITS and trnL–trnF IGS sequence data. Plant Syst. Evol. 2010, 286, 39–58. [Google Scholar]
- Yang, Y.; Berry, P.E. Phylogenetics of the Chamaesyce clade (Euphorbia, Euphorbiaceae): Reticulate evolution and long-distance dispersal in a prominent C4 lineage. Am. J. Bot. 2011, 98, 1486–1503. [Google Scholar] [CrossRef]
- Genc, I.; Kültür, Ş. Euphorbia akmanii (Euphorbiaceae), a new species from Turkey. Phytotaxa 2016, 265, 112–120. [Google Scholar] [CrossRef]
- Ernst, M.; Nothias, L.-F.; van der Hooft, J.J.; Silva, R.R.; Saslis-Lagoudakis, C.H.; Grace, O.M.; Martinez-Swatson, K.; Hassemer, G.; Funez, L.A.; Simonsen, H.T. Assessing specialized metabolite diversity in the cosmopolitan plant genus Euphorbia, L. Front. Plant Sci. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.; Kamla, P. Phytoconstituents of Euphorbia species. Fitoterapia 1990, 41, 483–516. [Google Scholar]
- Abdelgaleil, S.A.; Kassem, S.M.; Doe, M.; Baba, M.; Nakatani, M. Diterpenoids from Euphorbia paralias. Phytochemistry 2001, 58, 1135–1139. [Google Scholar] [CrossRef]
- Ravikanth, V.; Reddy, V.N.; Rao, T.P.; Diwan, P.; Ramakrishna, S.; Venkateswarlu, Y. Macrocyclic diterpenes from Euphorbia nivulia. Phytochemistry 2002, 59, 331–335. [Google Scholar] [CrossRef]
- Ernst, M.; Grace, O.M.; Saslis-Lagoudakis, C.H.; Nilsson, N.; Simonsen, H.T.; Rønsted, N. Global medicinal uses of Euphorbia, L.(Euphorbiaceae). J. Ethnopharmacol. 2015, 176, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, M.; Ali, M.; Tala, M.F.; Hamed, A.; Hassan, A.Z. Ecological and Phytochemical Studies on Euphorbia retusa (Forssk.) from Egyptian Habitat. J. Anal. Methods Chem. 2018, 2018, 9143683. [Google Scholar] [CrossRef]
- Rahman, A.H.M.M.; Akter, M. Taxonomy and Medicinal Uses of Euphorbiaceae (Spurge) Family of Rajshahi, Bangladesh. Res. Plant Sci. 2013, 1, 74–80. [Google Scholar]
- Kumar, S.; Malhotra, R.; Kumar, D. Euphorbia hirta: Its chemistry, traditional and medicinal uses, and pharmacological activities. Pharmacogn. Rev. 2010, 4, 58. [Google Scholar] [CrossRef]
- Al-Mahmooli, I.; Al-Bahri, Y.; Al-Sadi, A.; Deadman, M. First report of Euphorbia larica dieback caused by Fusarium brachygibbosum in Oman. Plant Dis. 2013, 97, 687. [Google Scholar] [CrossRef]
- Miller, A.G.; Morris, M. Plants of Dhofar: The Southern Region of Oman, Traditional, Economic and Medicinal Uses; Oman: Office of the Adviser for Conservation of the Environment; Diwan of Royal Court: Sultanate, Oman, 1988; ISBN 715708082. [Google Scholar]
- Noori, M.; Chehreghani, A.; Kaveh, M. Flavonoids of 17 species of Euphorbia (Euphorbiaceae) in Iran. Toxicol. Environ. Chem. 2009, 91, 631–641. [Google Scholar] [CrossRef]
- Patzelt, A. Oman Plant: Red Data Book; Oman Botanic Garden: Muscat, Oman, 2015. [Google Scholar]
- Pickering, H.; Patzelt, A. Field Guide to the Wild Plants of Oman; Royal Botanic Gardens: Muscat, Oman, 2008. [Google Scholar]
- Trapnell, D.W.; Hamrick, J.; Negrón-Ortiz, V. Genetic diversity within a threatened, endemic North American species, Euphorbia telephioides (Euphorbiaceae). Conserv. Genet. 2012, 13, 743–751. [Google Scholar] [CrossRef]
- Trejo, L.; Briones-Dumas, E.; Gómez-Bermejo, R.; Olson, M.E. Molecular evidence for repeated recruitment of wild Christmas poinsettia (Euphorbia pulcherrima) into traditional horticulture in Mexico. Genet. Resour. Crop Evol. 2019, 66, 481–490. [Google Scholar] [CrossRef]
- Dorsey, B.L.; Haevermans, T.; Aubriot, X.; Morawetz, J.J.; Riina, R.; Steinmann, V.W.; Berry, P.E. Phylogenetics, morphological evolution, and classification of Euphorbia subgenus Euphorbia. Taxon 2013, 62, 291–315. [Google Scholar] [CrossRef]
- Asif, M.H.; Mantri, S.S.; Sharma, A.; Srivastava, A.; Trivedi, I.; Gupta, P.; Mohanty, C.S.; Sawant, S.V.; Tuli, R. Complete sequence and organisation of the Jatropha curcas (Euphorbiaceae) chloroplast genome. Tree Genet. Genomes 2010, 6, 941–952. [Google Scholar] [CrossRef]
- Khan, A.; Asaf, S.; Khan, A.L.; Al-Harrasi, A.; Al-Sudairy, O.; AbdulKareem, N.M.; Khan, A.; Shehzad, T.; Alsaady, N.; Al-Lawati, A.; et al. First complete chloroplast genomics and comparative phylogenetic analysis of Commiphora gileadensis and C. foliacea: Myrrh producing trees. PLoS ONE 2019, 14, e0208511. [Google Scholar] [CrossRef]
- Khan, A.; Asaf, S.; Khan, A.L.; Khan, A.; Al-Harrasi, A.; Al-Sudairy, O.; AbdulKareem, N.M.; Al-Saady, N.; Al-Rawahi, A. Complete chloroplast genomes of medicinally important Teucrium species and comparative analyses with related species from Lamiaceae. PeerJ 2019, 7, e7260. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Long, H.; Zhang, L.; Liu, Z.; Cao, H.; Shi, M.; Tan, X. The complete chloroplast genome sequence of tung tree (Vernicia fordii): Organization and phylogenetic relationships with other angiosperms. Sci. Rep. 2017, 7, 1869. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, H. Research progress of sugarcane chloroplast genome. Agric. Sci. Technol. 2013, 14, 1693. [Google Scholar]
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.-M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.-W.; Lee, I.-J. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef]
- Daniell, H.; Wurdack, K.J.; Kanagaraj, A.; Lee, S.-B.; Saski, C.; Jansen, R.K. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 2008, 116, 723. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qu, Z.; Tian, X. Complete chloroplast genome of an endangered oil tree, Deutzianthus tonkinensis (Euphorbiaceae). Mitochondrial DNA Part B 2019, 4, 299–300. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Zhao, L.; Duan, N.; Guo, H.-X.; Wang, C.-Y.; Liu, B.-B. Complete chloroplast genome of Euphorbia hainanensis (Euphorbiaceae), a rare cliff top boskage endemic to China. Mitochondrial DNA Part B 2019, 4, 1325–1326. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.e.; Li, C. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Yang, J.-B.; Tang, M.; Li, H.-T.; Zhang, Z.-R.; Li, D.-Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef]
- Do Nascimento Vieira, L.; Faoro, H.; Rogalski, M.; de Freitas Fraga, H.P.; Cardoso, R.L.A.; de Souza, E.M.; de Oliveira Pedrosa, F.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar]
- Khan, A.L.; Al-Harrasi, A.; Asaf, S.; Park, C.E.; Park, G.-S.; Khan, A.R.; Lee, I.-J.; Al-Rawahi, A.; Shin, J.-H. The first chloroplast genome sequence of Boswellia sacra, a resin-producing plant in Oman. PLoS ONE 2017, 12, e0169794. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete chloroplast genome of Tanaecium tetragonolobum: The first Bignoniaceae plastome. PLoS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Kim, K.-A.; Kwak, M.; Lee, B.; Yoo, K.-O. The complete chloroplast genome sequences of four Viola species (Violaceae) and comparative analyses with its congeneric species. PLoS ONE 2019, 14, e0214162. [Google Scholar] [CrossRef]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-S.; Yun, B.-K.; Yoon, Y.-H.; Hong, S.-Y.; Mekapogu, M.; Kim, K.-H.; Yang, T.-J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.-C.; Zhang, Y.-Z.; Geng, H.-M.; Chen, S.-L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Chung, M.G.; Park, S. The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): Comparative analysis and highly divergent regions. Front. Plant Sci. 2016, 7, 355. [Google Scholar] [CrossRef]
- Blazier, J.C.; Jansen, R.K.; Mower, J.P.; Govindu, M.; Zhang, J.; Weng, M.-L.; Ruhlman, T.A. Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 2016, 117, 1209–1220. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Wicke, S.; Wang, H.; Jin, J.-J.; Chen, S.-Y.; Zhang, S.-D.; Li, D.-Z.; Yi, T.-S. Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Front. Plant Sci. 2018, 9, 138. [Google Scholar] [CrossRef]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; McLenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of C olocasia esculenta (L.) S chott (A raceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef]
- Firetti, F.; Zuntini, A.R.; Gaiarsa, J.W.; Oliveira, R.S.; Lohmann, L.G.; Van Sluys, M.A. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.-F.; Li, Z.-H. Comparative analysis of the complete chloroplast genome of four endangered herbals of Notopterygium. Genes 2017, 8, 124. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Chaw, S.-M. Evolutionary stasis in cycad plastomes and the first case of plastome GC-biased gene conversion. Genome Biol. Evol. 2015, 7, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Peirson, J.A.; Bruyns, P.V.; Riina, R.; Morawetz, J.J.; Berry, P.E. A molecular phylogeny and classification of the largely succulent and mainly African Euphorbia subg. Athymalus (Euphorbiaceae). Taxon 2013, 62, 1178–1199. [Google Scholar] [CrossRef]
- Shi, C.; Hu, N.; Huang, H.; Gao, J.; Zhao, Y.-J.; Gao, L.-Z. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 2012, 7, e31468. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.; Khan, A.L.; Al-Harrasi, A.; Al-Rawahi, A. Complete Chloroplast Genomes of Vachellia nilotica and Senegalia senegal: Comparative Genomics and Phylogenomic Placement in a New Generic System. PloS oNE 2019, 14. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, L.; Beszteri, B.; Gäbler-Schwarz, S.; Held, C.; Leese, F.; Mayer, C.; Pöhlmann, K.; Frickenhaus, S. S TAMP: Extensions to the S TADEN sequence analysis package for high throughput interactive microsatellite marker design. BMC Bioinform. 2009, 10, 41. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D. PAUP-a computer-program for phylogenetic inference using maximum parsimony. J. Gen. Physiol. 1993, 102, A9. [Google Scholar]
- Wu, Z.; Tembrock, L.R.; Ge, S. Are differences in genomic data sets due to true biological variants or errors in genome assembly: An example from two chloroplast genomes. PLoS ONE 2015, 10, e0118019. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.-M.; Khan, M.A.; Lee, S.-M.; Lee, I.-J. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 2016, 7, 843. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. smithii | E. larica | E. esula | E. tirucalli | M. esculanta | J. curcas | H. brasiliensis | R. communis | V. fordii | C. tiglium | D. tonkinensis | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | 162,172 | 162,358 | 160,512 | 163,091 | 161,453 | 163,856 | 161,191 | 163,161 | 161,528 | 150,021 | 163,481 |
| Overall GC contents | 35.8 | 35.6 | 35.6 | 35.6 | 35.9 | 35.4 | 35.7 | 35.7 | 36.0 | 35.4 | 35.7 |
| LSC size in bp | 91,158 | 91,537 | 90,309 | 91,259 | 89,275 | 91,846 | 89,209 | 89,650 | 89,132 | 111,654 | 91,453 |
| SSC size in bp | 18,603 | 18,239 | 17,023 | 18,168 | 18,250 | 17,849 | 18,362 | 18,816 | 18,758 | 18,167 | 18,476 |
| IR size in bp | 26,206 | 26,291 | 26,590 | 26,832 | 26,954 | 27,023 | 26,810 | 27,347 | 26,819 | 10,100 | 26,776 |
| Protein coding regions size in bp | 79,173 | 80,458 | 80,274 | 74,289 | 72,108 | 79,206 | 78,852 | 79,494 | 80,283 | 68,601 | 79,857 |
| tRNA size in bp | 2885 | 2887 | 2925 | 2885 | 2814 | 2797 | 2812 | 2802 | 2742 | 2560 | 2740 |
| rRNA size in bp | 9050 | 9049 | 9049 | 9049 | 6252 | 9047 | 9050 | 9050 | 9048 | 9050 | 9050 |
| Number of genes | 132 | 133 | 132 | 130 | 128 | 129 | 129 | 131 | 129 | 122 | 134 |
| Number of protein coding genes | 85 | 86 | 85 | 82 | 83 | 84 | 84 | 86 | 85 | 78 | 84 |
| Number of rRNA | 8 | 8 | 8 | 8 | 7 | 8 | 8 | 8 | 8 | 8 | 8 |
| Number of tRNA | 39 | 39 | 39 | 39 | 38 | 37 | 37 | 37 | 36 | 34 | 36 |
| Genes with introns | 19 | 19 | 18 | 13 | 20 | 21 | 21 | 19 | 21 | 20 | 20 |
| Gene | Exon I (bp) | Intron 1 (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | |
| atpF | 145 | 145 | 145 | 145 | 670 | 671 | 666 | 667 | 470 | 470 | 470 | 470 | ||||||||
| petB | 6 | 6 | 773 | 779 | 642 | 642 | ||||||||||||||
| PetD | 8 | 8 | 779 | 780 | 496 | 496 | ||||||||||||||
| rpl2* | 400 | 396 | 396 | 396 | 634 | 629 | 624 | 629 | 464 | 468 | 468 | 468 | ||||||||
| rpl16 | 9 | 9 | 1395 | 1395 | 399 | 399 | ||||||||||||||
| rpoC1 | 430 | 432 | 432 | 767 | 769 | 775 | 1613 | 1618 | 1617 | |||||||||||
| rps12* | 114 | 114 | 114 | 114 | 232 | 232 | 232 | 232 | 541 | 536 | 536 | 536 | 26 | 26 | 26 | 26 | ||||
| clpP | 71 | 71 | 71 | 71 | 825 | 831 | 821 | 827 | 291 | 291 | 291 | 291 | 650 | 648 | 653 | 651 | 229 | 229 | 229 | 229 |
| ndhA | 553 | 554 | 553 | 553 | 1138 | 1111 | 1116 | 1137 | 539 | 541 | 539 | 539 | ||||||||
| ndhB* | 777 | 777 | 777 | 682 | 682 | 678 | 756 | 756 | 756 | |||||||||||
| ycf3 | 124 | 124 | 124 | 124 | 733 | 747 | 747 | 733 | 230 | 230 | 230 | 230 | 669 | 676 | 675 | 677 | 153 | 153 | 153 | 153 |
| trnA-UGC* | 38 | 38 | 38 | 38 | 813 | 803 | 813 | 803 | 35 | 35 | 35 | 35 | ||||||||
| trnI-GAU* | 42 | 42 | 42 | 42 | 945 | 945 | 945 | 945 | 35 | 35 | 35 | 35 | ||||||||
| trnL-UAA | 37 | 37 | 37 | 37 | 587 | 583 | 619 | 590 | 50 | 50 | 50 | 50 | ||||||||
| trnK-UUU | 37 | 37 | 37 | 37 | 2551 | 2560 | 2555 | 2563 | 28 | 29 | 29 | 29 | ||||||||
| trnV-UAC | 39 | 582 | 42 | |||||||||||||||||
| T/U | C | A | G | Length (bp) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | E.l | E.s | E.e | E.t | |
| Genome | 32.7 | 32.5 | 32.7 | 32.6 | 18.1 | 18.1 | 18 | 18.1 | 31.7 | 31.7 | 31.8 | 31.8 | 17.5 | 17.6 | 17.6 | 17.5 | 162,258 | 162,172 | 160,512 | 163,091 |
| LSC | 34.5 | 34.3 | 34.5 | 34.3 | 16.7 | 16.8 | 16.6 | 16.7 | 32.8 | 32.7 | 32.9 | 32.9 | 16.0 | 16.1 | 16.1 | 16.1 | 91,537 | 91,158 | 90,309 | 91,259 |
| SSC | 35.0 | 34.6 | 34.8 | 35 | 15.7 | 15.8 | 15.9 | 15.8 | 34.9 | 34.9 | 35 | 34.9 | 14.3 | 14.7 | 14.3 | 14.3 | 18,239 | 18,603 | 17,023 | 1818 |
| IR | 28.9 | 29 | 28.7 | 29.1 | 20.5 | 20.5 | 21.9 | 20.4 | 28.6 | 28.5 | 29 | 28.6 | 22.0 | 22.1 | 20.4 | 21.9 | 26,291 | 26,206 | 26,590 | 26,832 |
| tRNA | 26.5 | 25.5 | 25.4 | 25.5 | 21.6 | 23.2 | 23.2 | 23.3 | 24.7 | 22.2 | 22.3 | 22.1 | 27.2 | 29.2 | 29.1 | 29.1 | 2887 | 2885 | 2925 | 2885 |
| rRNA | 18.8 | 18.8 | 18.8 | 18.8 | 23.7 | 23.7 | 23.7 | 23.7 | 25.8 | 25.8 | 25.7 | 25.7 | 31.8 | 31.8 | 31.8 | 31.8 | 2049 | 9050 | 9049 | 9049 |
| Protein Coding genes | 31.8 | 31.8 | 31.8 | 31.6 | 17.4 | 17.4 | 17.3 | 17.3 | 31.1 | 31.1 | 31.2 | 31.1 | 19.8 | 19.8 | 19.6 | 19.9 | 80,458 | 79,173 | 80,274 | 74,289 |
| 1st position | 24.3 | 25.9 | 24.1 | 23.9 | 18.3 | 18.4 | 18.4 | 18.5 | 31.0 | 31.2 | 31.0 | 30.9 | 26.1 | 24.3 | 26.3 | 26.6 | 26,817 | 26,390 | 26,758 | 24,763 |
| 2nd position | 32.6 | 32.7 | 28.4 | 38.4 | 19.9 | 19.8 | 19.9 | 19.9 | 29.7 | 29.8 | 29.8 | 29.9 | 17.5 | 17.5 | 17.4 | 17.5 | 26,817 | 26,390 | 26,758 | 24,763 |
| 3rd position | 38.2 | 38.2 | 38.6 | 38.4 | 32.2 | 13.8 | 16.6 | 13.4 | 13.8 | 32.1 | 28.4 | 32.4 | 15.5 | 18.1 | 13.8 | 15.7 | 26,817 | 26,390 | 26,758 | 24,763 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Asaf, S.; Khan, A.L.; Shehzad, T.; Al-Rawahi, A.; Al-Harrasi, A. Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny. Plants 2020, 9, 199. https://doi.org/10.3390/plants9020199
Khan A, Asaf S, Khan AL, Shehzad T, Al-Rawahi A, Al-Harrasi A. Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny. Plants. 2020; 9(2):199. https://doi.org/10.3390/plants9020199
Chicago/Turabian StyleKhan, Arif, Sajjad Asaf, Abdul Latif Khan, Tariq Shehzad, Ahmed Al-Rawahi, and Ahmed Al-Harrasi. 2020. "Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny" Plants 9, no. 2: 199. https://doi.org/10.3390/plants9020199
APA StyleKhan, A., Asaf, S., Khan, A. L., Shehzad, T., Al-Rawahi, A., & Al-Harrasi, A. (2020). Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny. Plants, 9(2), 199. https://doi.org/10.3390/plants9020199