New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.)

 ,
, 
Abstract
1. Introduction
2. Results
2.1. Bioinformatic Analysis
2.2. Marker Testing
2.3. Marker Polymorphism, Transferability, and Reproducibility
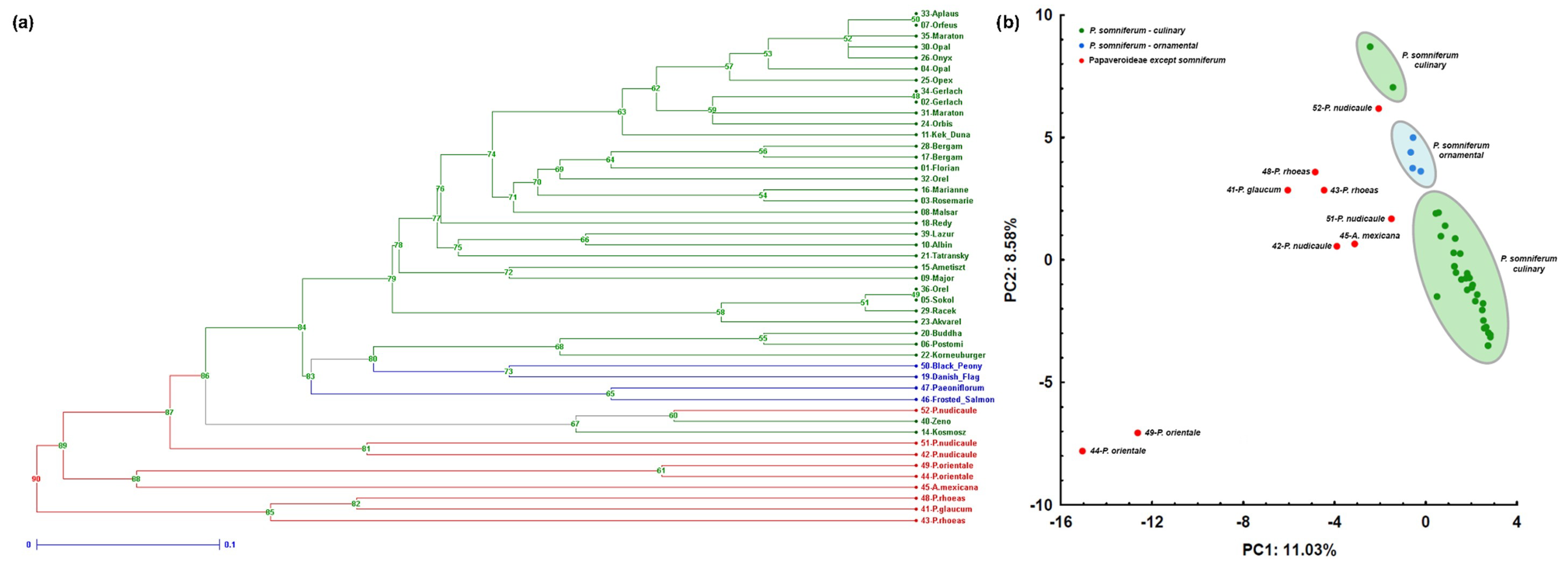
2.4. Assessment of Genetic Diversity and Discrimination Power of Markers
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Bioinformatic Analysis and Primers Design
4.3. SSR Loci Amplification
4.4. Fragment Analysis
4.5. Sequencing
4.6. Statistical Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stranska, I.; Skalicky, M.; Novak, J.; Matyasova, E.; Hejnak, V. Analysis of selected poppy (Papaver somniferum L.) cultivars: Pharmaceutically important alkaloids. Ind. Crop. Prod. 2013, 41, 120–126. [Google Scholar] [CrossRef]
- Coward, F.; Shennan, S.; Colledge, S.; Conolly, J.; Collard, M. The spread of Neolithic plant economies from the Near East to northwest Europe: A phylogenetic analysis. J. Archaeol. Sci. 2008, 35, 42–56. [Google Scholar] [CrossRef]
- Merlin, M.D. Archaeological evidence for the tradition of psychoactive plant use in the old world. Econ. Bot. 2003, 57, 295–323. [Google Scholar] [CrossRef]
- Kabera, J.N.; Semana, E.; Mussa, A.R.; He, X. Plant secondary metabolites: Biosynthesis, classification, function and pharmacological properties. J. Pharm. Pharm. 2014, 2, 377–392. [Google Scholar]
- Winzer, T.; Gazda, V.; He, Z.; Kaminski, F.; Kern, M.; Larson, T.R.; Li, Y.; Meade, F.; Teodor, R.; Vaistij, F.E.; et al. A Papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science 2012, 336, 1704–1708. [Google Scholar] [CrossRef]
- Marciano, M.A.; Panicker, S.X.; Liddil, G.D.; Lindgren, D.; Sweder, K.S. Development of a method to extract opium poppy (Papaver somniferum L.) DNA from heroin. Sci. Rep. 2018, 8, 2590. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Sproll, C.; Musshoff, F. Poppy seed foods and opiate drug testing-where are we today? Ther. Drug. Monit. 2010, 32, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Jankovičová, K.; Ulbrich, P.; Fuknová, M. Effect of poppy seed consummation on the positive results of opiates screening in biological samples. Leg. Med. 2009, 11, S416–S418. [Google Scholar] [CrossRef] [PubMed]
- Lavania, U.C.; Srivastava, S. Quantitative delineation of karyotype variation in Papaver as a measure of phylogenetic differentiation and origin. Curr. Sci. 1999, 77, 7. [Google Scholar]
- Tétényi, P. Opium Poppy (Papaver somniferum): Botany and Horticulture. In Horticultural Reviews; Janick, J., Ed.; John Wiley & Sons, Inc.: Oxford, UK, 2010; pp. 373–408. ISBN 978-0-470-65062-2. [Google Scholar]
- Labanca, F.; Ovesnà, J.; Milella, L. Papaver somniferum L. taxonomy, uses and new insight in poppy alkaloid pathways. Phytochem. Rev. 2018, 17, 853–871. [Google Scholar] [CrossRef]
- Choe, S.; Kim, S.; Lee, C.; Yang, W.; Park, Y.; Choi, H.; Chung, H.; Lee, D.; Hwang, B.Y. Species identification of Papaver by metabolite profiling. Forensic Sci. Int. 2011, 211, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.; Baranska, M.; Quilitzsch, R.; Schütze, W. Determination of alkaloids in capsules, milk and ethanolic extracts of poppy (Papaver somniferum L.) by ATR-FT-IR and FT-Raman spectroscopy. Analyst 2004, 129, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Odell, L.R.; Skopec, J.; McCluskey, A. Isolation and identification of unique marker compounds from the Tasmanian poppy Papaver somniferum N. Forensic Sci. Int. 2008, 175, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Németh-Zámbori, É.; Jászberényi, C.; Rajhárt, P.; Bernáth, J. Evaluation of alkaloid profiles in hybrid generations of different poppy (Papaver somniferum L.) genotypes. Ind. Crop. Prod. 2011, 33, 690–696. [Google Scholar] [CrossRef]
- Gümüşçü, A.; Arslan, N.; Sarıhan, E.O. Evaluation of selected poppy (Papaver somniferum L.) lines by their morphine and other alkaloids contents. Eur. Food Res. Technol. 2008, 226, 1213–1220. [Google Scholar] [CrossRef]
- Dittbrenner, A.; Mock, H.P.; Börner, A.; Lohwasser, U. Variability of alkaloid content in Papaver somniferum L. J. Appl. Bot. Food Qual. 2009, 82, 103–107. [Google Scholar]
- Ziegler, J.; Facchini, P.J.; Geißler, R.; Schmidt, J.; Ammer, C.; Kramell, R.; Voigtländer, S.; Gesell, A.; Pienkny, S.; Brandt, W. Evolution of morphine biosynthesis in opium poppy. Phytochemistry 2009, 70, 1696–1707. [Google Scholar] [CrossRef]
- Güçlü, G.B.; Gürkök, T.; Koyuncu, M.; Arslan, N.; Parmaksız, İ. Genetic characterization of Turkish commercial opium poppy (Papaver somniferum L.) cultivars using ISSR and SSR markers. J. New Results Sci. 2014, 3, 48–57. [Google Scholar]
- Parmaksiz, İ.; Özcan, S. Morphological, chemical, and molecular analyses of Turkish papaver accessions (Sect. Oxytona). Turk. J. Bot. 2011, 35, 1–16. [Google Scholar]
- Acharya, H.S.; Sharma, V. Molecular characterization of opium poppy (Papaver somniferum) germplasm. Am. J. Infect. Dis. 2009, 5, 155–160. [Google Scholar] [CrossRef]
- Saunders, J.A.; Pedroni, M.J.; Penrose, L.D.J.; Fist, A.J. AFLP analysis of opium poppy. Crop. Sci. 2001, 41, 1596. [Google Scholar] [CrossRef]
- Mičianová, V.; Ondreičková, K.; Muchová, D.; Klčová, L.; Hudcovicová, M.; Havrlentová, M.; Mihálik, D.; Kraic, J. Forensic application of EST-derived STR markers in opium poppy. Biologia 2017, 72, 587–594. [Google Scholar] [CrossRef]
- Celik, I.; Gultekin, V.; Allmer, J.; Doganlar, S.; Frary, A. Development of genomic simple sequence repeat markers in opium poppy by next-generation sequencing. Mol. Breed. 2014, 34, 323–334. [Google Scholar] [CrossRef]
- Şelale, H.; Çelik, I.; Gültekin, V.; Allmer, J.; Doğanlar, S.; Frary, A. Development of EST-SSR markers for diversity and breeding studies in opium poppy. Plant. Breed. 2013, 132, 344–351. [Google Scholar] [CrossRef]
- Lee, E.J.; Jin, G.N.; Lee, K.L.; Han, M.S.; Lee, Y.H.; Yang, M.S. Exploiting expressed sequence tag databases for the development and characterization of gene-derived simple sequence repeat markers in the opium poppy (Papaver somniferum L.) for forensic applications. J. Forensic Sci. 2011, 56, 1131–1135. [Google Scholar] [CrossRef]
- Richard, G.-F.; Kerrest, A.; Dujon, B. Comparative Genomics and Molecular Dynamics of DNA Repeats in Eukaryotes. Microbiol. Mol. Biol. Rev. 2008, 72, 686–727. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Buschiazzo, E.; Gemmell, N.J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Parida, S.K.; Kalia, S.K.; Kaul, S.; Dalal, V.; Hemaprabha, G.; Selvi, A.; Pandit, A.; Singh, A.; Gaikwad, K.; Sharma, T.R.; et al. Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor. Appl. Genet. 2009, 118, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.D.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.C.; Tollenaere, R.; Dalton-Morgan, J.; Batley, J. Molecular Marker Applications in Plants. In Plant Genotyping; Batley, J., Ed.; Springer: New York, NY, USA, 2015; Volume 1245, pp. 13–27. ISBN 978-1-4939-1965-9. [Google Scholar]
- Meyer, L.; Causse, R.; Pernin, F.; Scalone, R.; Bailly, G.; Chauvel, B.; Délye, C.; Le Corre, V. New gSSR and EST-SSR markers reveal high genetic diversity in the invasive plant Ambrosia artemisiifolia L. and can be transferred to other invasive Ambrosia species. PLoS ONE 2017, 12, e0176197. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Liu, X.; Hu, L.; Wang, S.; Cheng, X. De novo transcriptomic analysis of cowpea (Vigna unguiculata L. Walp.) for genic SSR marker development. BMC Genet. 2017, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- El-Esawi, M.A.; Germaine, K.; Bourke, P.; Malone, R. Genetic diversity and population structure of Brassica oleracea germplasm in Ireland using SSR markers. C. R. Biol. 2016, 339, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kishine, M.; Tsutsumi, K.; Kitta, K. A set of tetra-nucleotide core motif SSR markers for efficient identification of potato (Solanum tuberosum) cultivars. Breed. Sci. 2017, 67, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Bora, A.; Choudhury, P.R.; Pande, V.; Mandal, A.B. Assessment of genetic purity in rice (Oryza sativa L.) hybrids using microsatellite markers. 3 Biotech 2016, 6, 50. [Google Scholar] [CrossRef]
- Subirana, J.A.; Messeguer, X. Structural families of genomic microsatellites. Gene 2008, 408, 124–132. [Google Scholar] [CrossRef]
- Lai, Y.; Sun, F. The relationship between microsatellite slippage mutation rate and the number of repeat units. Mol. Biol. Evol. 2003, 20, 2123–2131. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [PubMed]
- Van de Peer, Y.; Fawcett, J.A.; Proost, S.; Sterck, L.; Vandepoele, K. The flowering world: A tale of duplications. Trends Plant. Sci. 2009, 14, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Schemske, D.W. Pathway, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef]
- Kati, V.; Le Corre, V.; Michel, S.; Jaffrelo, L.; Poncet, C.; Délye, C. Isolation and characterisation of 11 polymorphic microsatellite markers in Papaver rhoeas L. (corn poppy), a major annual plant species from cultivated areas. Int. J. Mol. Sci. 2012, 14, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef]
- Baldoni, L.; Cultrera, N.G.; Mariotti, R.; Ricciolini, C.; Arcioni, S.; Vendramin, G.G.; Buonamici, A.; Porceddu, A.; Sarri, V.; Ojeda, M.A.; et al. A consensus list of microsatellite markers for olive genotyping. Mol. Breed. 2009, 24, 213–231. [Google Scholar] [CrossRef]
- Drašnarová, A.; Krak, K.; Vít, P.; Doudová, J.; Douda, J.; Hadincová, V.; Zákravský, P.; Mandák, B. Cross-amplification and multiplexing of SSR markers for Alnus glutinosa and A. incana. Tree Genet. Genomes 2014, 10, 865–873. [Google Scholar] [CrossRef]
- Kameníková, L.; Vrbovský, V. White-seeded poppy varieties Orel and Racek. Czech. J. Genet. Plant. Breed. 2009, 45, 37–38. [Google Scholar] [CrossRef]
- Butler, J.M. Advanced Topics in Forensic DNA Typing: Interpretation; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef]
- Meier, U. BBCH-Monograph: Growth Stages of Mono-and Dicotyledonous Plants; Blackwell Wissenschafts-Verlag: Berlin, Germany, 1997. [Google Scholar]
- Masoudi-Nejad, A.; Tonomura, K.; Kawashima, S.; Moriya, Y.; Suzuki, M.; Itoh, M.; Kanehisa, M.; Endo, T.; Goto, S. EGassembler: Online bioinformatics service for large-scale processing, clustering and assembling ESTs and genomic DNA fragments. Nucleic Acids Res. 2006, 34, W459–W462. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant. Sci. 2016, 7, 1350. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Proceedings of the Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- TIBCO Statistica. Available online: https://www.tibco.com/resources/product-download/tibco-statistica-trial-download-windows (accessed on 28 December 2019).
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. Available online: http://darwin.cirad.fr/darwin (accessed on 26 April 2019).
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Hintze, J. NCSS; NCSS: Kaysville, UT, USA, 2001. [Google Scholar]


{kind=link}
| Nr. of Tested Markers | Amplifying Markers | Useful Markers | Total Nr. of Alleles*/Fragments** | Mean per Locus (Papaveroideae) | Mean per Locus (P. somniferum) | Transferability a (%) | Study |
|---|---|---|---|---|---|---|---|
| 100 | 96 | 53 | 207 ** | 3.9 | 1.7 | 97 | [24] |
| 93 | 76 | 67 | 562 ** | 8.4 | 3.77 | 88.7 | [25] |
| 22 | 17 | 6 | 17 * | - | 2.83 | 33 | [26] |
| 12 | 12 | 8 | 20 * | - | 2.5 | - | [23] |
| 203 | 166 | 17 | 118 * | 6.94 | 5.18 | 29.41 | this study |
| Marker | All Accessions | P. somniferum | P. somniferum-Culinary | ||||||
|---|---|---|---|---|---|---|---|---|---|
| k | HO | PIC | k | HO | PIC | k | HO | PIC | |
| OPEST026 | 10 | 0.190 | 0.724 | 8 | 0.158 | 0.682 | 7 | 0.176 | 0.662 |
| OPEST048c | 9 | 0.114 | 0.599 | 7 | 0.053 | 0.536 | 6 | 0.029 | 0.450 |
| OPEST051c | 5 | 0.077 | 0.538 | 5 | 0.079 | 0.517 | 5 | 0.088 | 0.485 |
| OPEST053c | 10 | 0.109 | 0.674 | 6 | 0.053 | 0.598 | 6 | 0.059 | 0.551 |
| OPEST061 | 7 | 0.119 | 0.694 | 6 | 0.132 | 0.672 | 6 | 0.118 | 0.654 |
| OPEST081c | 8 | 0.190 | 0.559 | 6 | 0.158 | 0.526 | 5 | 0.147 | 0.503 |
| OPEST086d | 3 | 0.068 | 0.245 | 3 | 0.026 | 0.164 | 2 | 0.029 | 0.081 |
| OPEST099 | 8 | 0.109 | 0.590 | 3 | 0.053 | 0.428 | 3 | 0.029 | 0.425 |
| OPEST102b | 9 | 0.087 | 0.505 | 5 | 0.053 | 0.371 | 4 | 0.029 | 0.344 |
| OPEST106 | 6 | 0.093 | 0.703 | 4 | 0.079 | 0.631 | 4 | 0.059 | 0.615 |
| OPEST120b | 7 | 0.159 | 0.605 | 3 | 0.105 | 0.478 | 3 | 0.088 | 0.432 |
| OPEST126b | 6 | 0.049 | 0.501 | 5 | 0.053 | 0.422 | 3 | 0.029 | 0.381 |
| OPEST131 | 6 | 0.111 | 0.518 | 4 | 0.105 | 0.399 | 4 | 0.088 | 0.418 |
| OPEST156 | 3 | 0.053 | 0.366 | 3 | 0.053 | 0.366 | 3 | 0.029 | 0.368 |
| OPEST169 | 6 | 0.075 | 0.531 | 5 | 0.079 | 0.517 | 2 | 0.088 | 0.485 |
| OPEST177b | 6 | 0.048 | 0.349 | 6 | 0.053 | 0.228 | 5 | 0.000 | 0.105 |
| OPGSSR001 | 9 | 0.154 | 0.693 | 9 | 0.158 | 0.699 | 9 | 0.088 | 0.654 |
| mean | 6.94 | 0.104 | 0.564 | 5.18 | 0.084 | 0.500 | 4.53 | 0.068 | 0.466 |
| median | 7 | 0.101 | 0.575 | 5 | 0.066 | 0.517 | 4 | 0.059 | 0.468 |
| Accession Number | Assigned Code | Species | Genebank Evidence Number | Variety | Source |
|---|---|---|---|---|---|
| 1 | FLO | P. somniferum | 15O0800171 | Florian | genebank Opava a |
| 2 | GER | P. somniferum | 15O0800148 | Gerlach | genebank Opava a |
| 3 | ROS | P. somniferum | 15O0800164 | Rosemarie | genebank Opava a |
| 4 | OPA | P. somniferum | 15O0800169 | Opal | genebank Opava a |
| 5 | SOK | P. somniferum | 15O0800179 | Sokol | genebank Opava a |
| 6 | POS | P. somniferum | 15O0800195 | Postomi | genebank Opava a |
| 7 | ORF | P. somniferum | 15O0800190 | Orfeus | genebank Opava a |
| 8 | MAL | P. somniferum | 15O0800183 | Malsar | genebank Opava a |
| 9 | MAJ | P. somniferum | 15O0800182 | Major | genebank Opava a |
| 10 | ALB | P. somniferum | 15O0800159 | Albín | genebank Opava a |
| 11 | KEK | P. somniferum | 15O0800093 | Kék Duna | genebank Opava a |
| 14 | KOS | P. somniferum | 15O0800173 | Kozmosz | genebank Opava a |
| 15 | AME | P. somniferum | 15O0800198 | Ametiszt | genebank Opava a |
| 16 | MAE | P. somniferum | 15O0800031 | Marianne | genebank Opava a |
| 17 | BER | P. somniferum | 15O0800184 | Bergam | genebank Opava a |
| 18 | RED | P. somniferum | 15O0800189 | Redy | genebank Opava a |
| 19 | DAN | P. somniferum | Danish Flag | market | |
| 20 | BUD | P. somniferum | 15O0800185 | Buddha | genebank Opava a |
| 21 | TAT | P. somniferum | Tatranský | Červený Újezd b | |
| 22 | KOR | P. somniferum | Korneuburger | Červený Újezd b | |
| 23 | AKV | P. somniferum | Akvarel | Červený Újezd b | |
| 24 | ORB_II | P. somniferum | Orbis | CISTA c | |
| 25 | OPE_II | P. somniferum | Opex | CISTA c | |
| 26 | ONY_II | P. somniferum | Onyx | CISTA c | |
| 28 | BER_II | P. somniferum | Bergam | CISTA c | |
| 29 | RAC_II | P. somniferum | Racek | CISTA c | |
| 30 | OPA_II | P. somniferum | Opal | CISTA c | |
| 31 | MAR | P. somniferum | 15O0800181 | Maraton | genebank Opava a |
| 32 | ORE | P. somniferum | 15O0800187 | Orel | genebank Opava a |
| 33 | APL_II | P. somniferum | Aplaus | CISTA c | |
| 34 | GER_II | P. somniferum | Gerlach | CISTA c | |
| 35 | MAR_II | P. somniferum | Maraton | CISTA c | |
| 36 | ORE_II | P. somniferum | Orel | CISTA c | |
| 39 | LAZ | P. somniferum | Lazur | Červený Újezd b | |
| 40 | ZEN | P. somniferum | Zeno | Červený Újezd b | |
| 41 | GLA | P. glaucum | seed mixture | market | |
| 42 | GNO | P. nudicaule | Gnome | market | |
| 43 | RHO | P. rhoeas | seed mixture | market | |
| 44 | PIZ | P. orientale | Pizziato | market | |
| 45 | MEX | A. mexicana | seed mixture | market | |
| 46 | FRO | P. somniferum | Frosted Salmon | market | |
| 47 | PAE | P. somniferum | Paeoniflorum | market | |
| 48 | DAW | P. rhoeas | Dawn Chorus | market | |
| 49 | SCHA | P. orientale | Scharlach | market | |
| 50 | PEO | P. somniferum | Black Peony | market | |
| 51 | NUD_I | P. nudicaule | seed mixture | market | |
| 52 | NUD_II | P. nudicaule | seed mixture | market |
| Marker | Dye | Primer sequence 5′- 3′ | Size range [bp] | GenBank AC Number | |
|---|---|---|---|---|---|
| OPEST026 | 6FAM | F- | GTGAGGAGGACGAGCTTTTG | 105–148 | MK744101 |
| R- | gtttcttCCGTTGTAAAATACCGACTGC | ||||
| OPEST048c | PET | F- | CGTGAGAAGCTAGAACAGAAAGA | 174–204 | MK744102 |
| R- | TCGTTCACTGAGTTCTGATATGA | ||||
| OPEST051c | VIC | F- | GGGTTCTTTTGTTCTACTTCTTTCTT | 147–162 | MK744103 |
| R- | AAGGTGTCGGTGCCCAGC | ||||
| OPEST053c | NED | F- | TCAATACCCACAAAAGGAGGA | 172–203 | MK744104 |
| R- | gtttcttTCAAGACAAAGAAACCAAGCCA | ||||
| OPEST061 | 6FAM | F- | GGCTGCTGCTTCTTTTCATC | 191–236 | MK744105 |
| R- | ATAGGGCAAACTGCCTGCTA | ||||
| OPEST081c | PET | F- | AGTAAAACGATCCGTACCTACCTGA | 133–176 | MK744106 |
| R- | CGTTTTTCTACAGGGTTGATTTCTGA | ||||
| OPEST086d | PET | F- | ACCTTTCCCCCTCTTCAGTAGC | 223–244 | MK744107 |
| R- | TCCAGTCCACATCAGGATCA | ||||
| OPEST099 | 6FAM | F- | TTAACAGATCCGCATTTCCA | 262–288 | MK744108 |
| R- | CACCGATTGTACCACGAAGA | ||||
| OPEST102b | VIC | F- | CGCCACCACATATTTCTCTG | 184–206 | MK744109 |
| R- | GGTTGTCGGCATAGAAGGAA | ||||
| OPEST106 | 6FAM | F- | CACCAAATCTCATTGCCTGA | 166–191 | MK744110 |
| R- | CCCTAATCGGATGGATCAAA | ||||
| OPEST120b | 6FAM | F- | TAGTGGTTGCTCGTAGCGTC | 138–156 | MK744111 |
| R- | TCACGGTTCTTCTATCATGGTG | ||||
| OPEST126b | 6FAM | F- | GTTTCTCACGGAGGGATTTG | 206–228 | MK744112 |
| R- | CCGTTTCCCAACTTCGTAGA | ||||
| OPEST131 | VIC | F- | GTTCCAAACCACCAACCACA | 224–250 | MK744113 |
| R- | TTGTGAGGCCCTAGAGAGGA | ||||
| OPEST156 | 6FAM | F- | TTTAGCTTACAATGGTGGGAGA | 264–270 | MK744114 |
| R- | GAAACCGTAGCCAGGTGAAA | ||||
| OPEST169 | VIC | F- | TCCAACGCAAGCAATTACAA | 165–205 | MK744115 |
| R- | GCCACTTCGTAACCCAGGTA | ||||
| OPEST177b | VIC | F- | TCTCCGTAACCTGAAGAACAGA | 96–112 | MK744116 |
| R- | TGGTGGCAGTGAATTTGAT | ||||
| OPGSSR001 | VIC | F- | TGCGGCTTCTAATCATCCTT | 218–244 | MK744117 |
| R- | CCATCAACTTCGCACAGCTA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vašek, J.; Čílová, D.; Melounová, M.; Svoboda, P.; Vejl, P.; Štikarová, R.; Vostrý, L.; Kuchtová, P.; Ovesná, J. New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.). Plants 2020, 9, 10. https://doi.org/10.3390/plants9010010
Vašek J, Čílová D, Melounová M, Svoboda P, Vejl P, Štikarová R, Vostrý L, Kuchtová P, Ovesná J. New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.). Plants. 2020; 9(1):10. https://doi.org/10.3390/plants9010010
Chicago/Turabian StyleVašek, Jakub, Daniela Čílová, Martina Melounová, Pavel Svoboda, Pavel Vejl, Radka Štikarová, Luboš Vostrý, Perla Kuchtová, and Jaroslava Ovesná. 2020. "New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.)" Plants 9, no. 1: 10. https://doi.org/10.3390/plants9010010
APA StyleVašek, J., Čílová, D., Melounová, M., Svoboda, P., Vejl, P., Štikarová, R., Vostrý, L., Kuchtová, P., & Ovesná, J. (2020). New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.). Plants, 9(1), 10. https://doi.org/10.3390/plants9010010