Gene Expression Maps in Plants: Current State and Prospects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
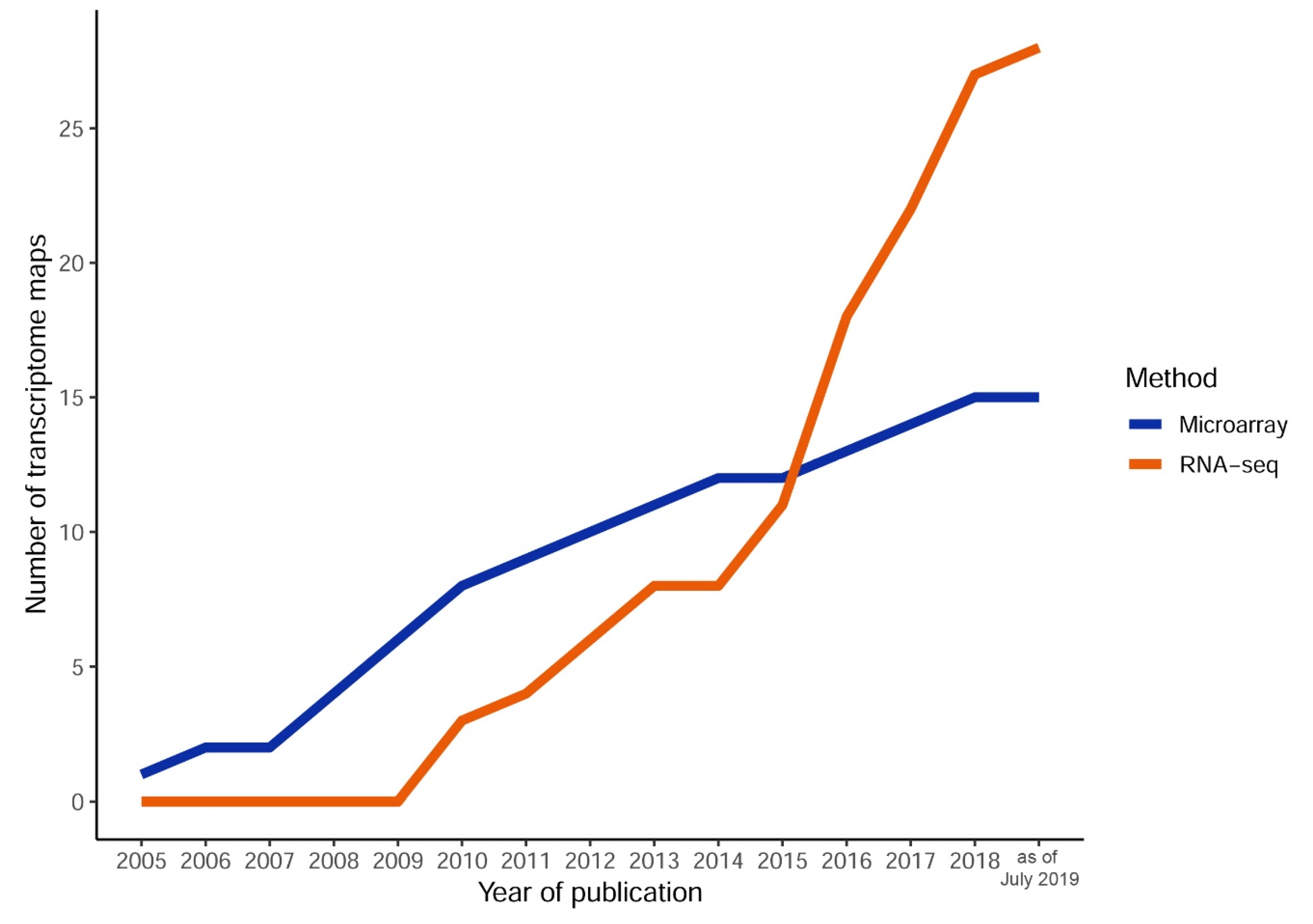
2. The Dawn of Plant Transcriptome Maps
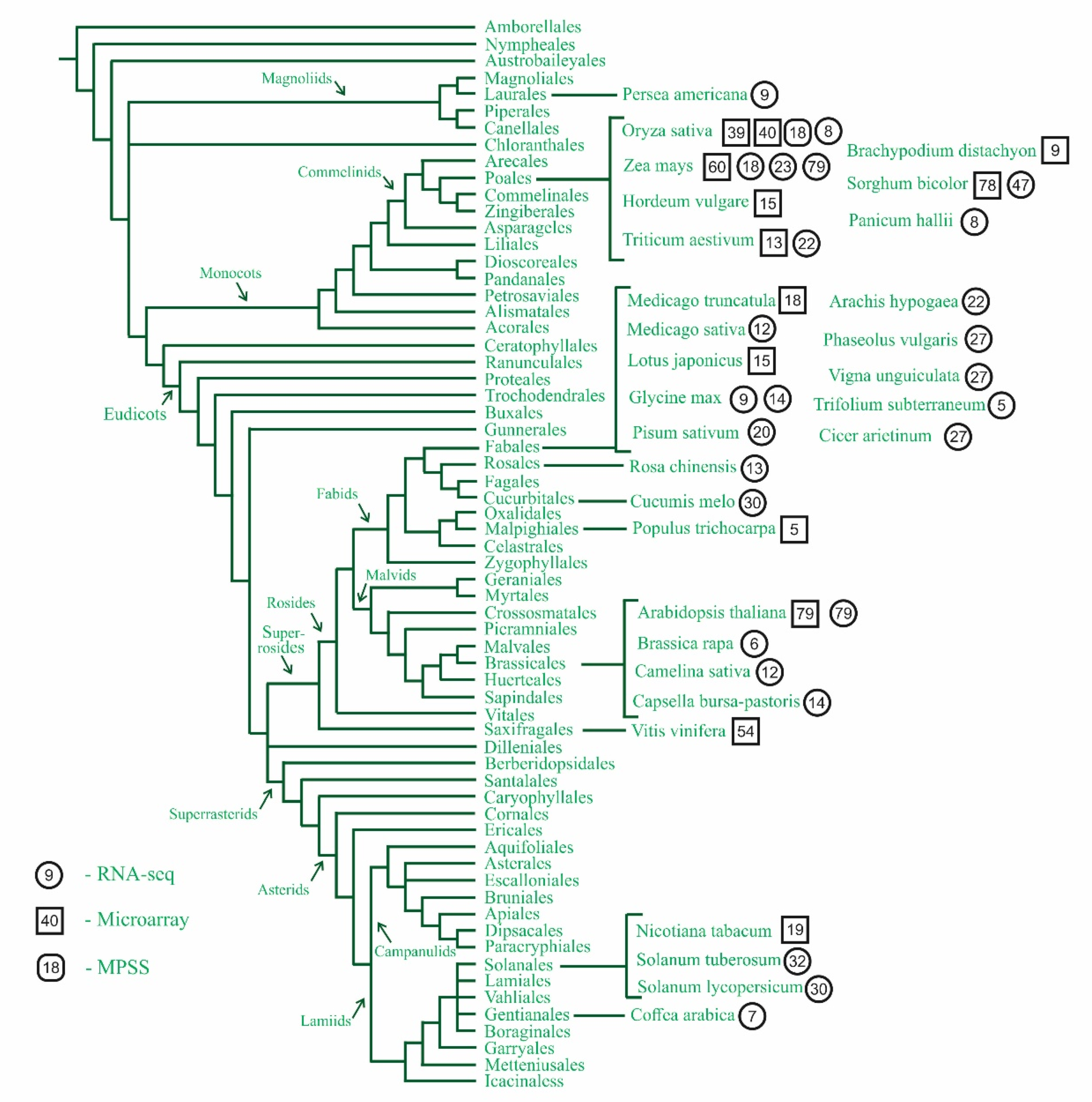
3. Where to Look for Uninvestigated Plants?
4. What Are the Modern Strategies of Transcriptome Map Construction?
5. How to Choose the Representative and Comparable Set of Samples?
6. Overall Characteristics of the Plant Transcriptome
7. What Questions Can Be Addressed by a Gene Expression Atlas?
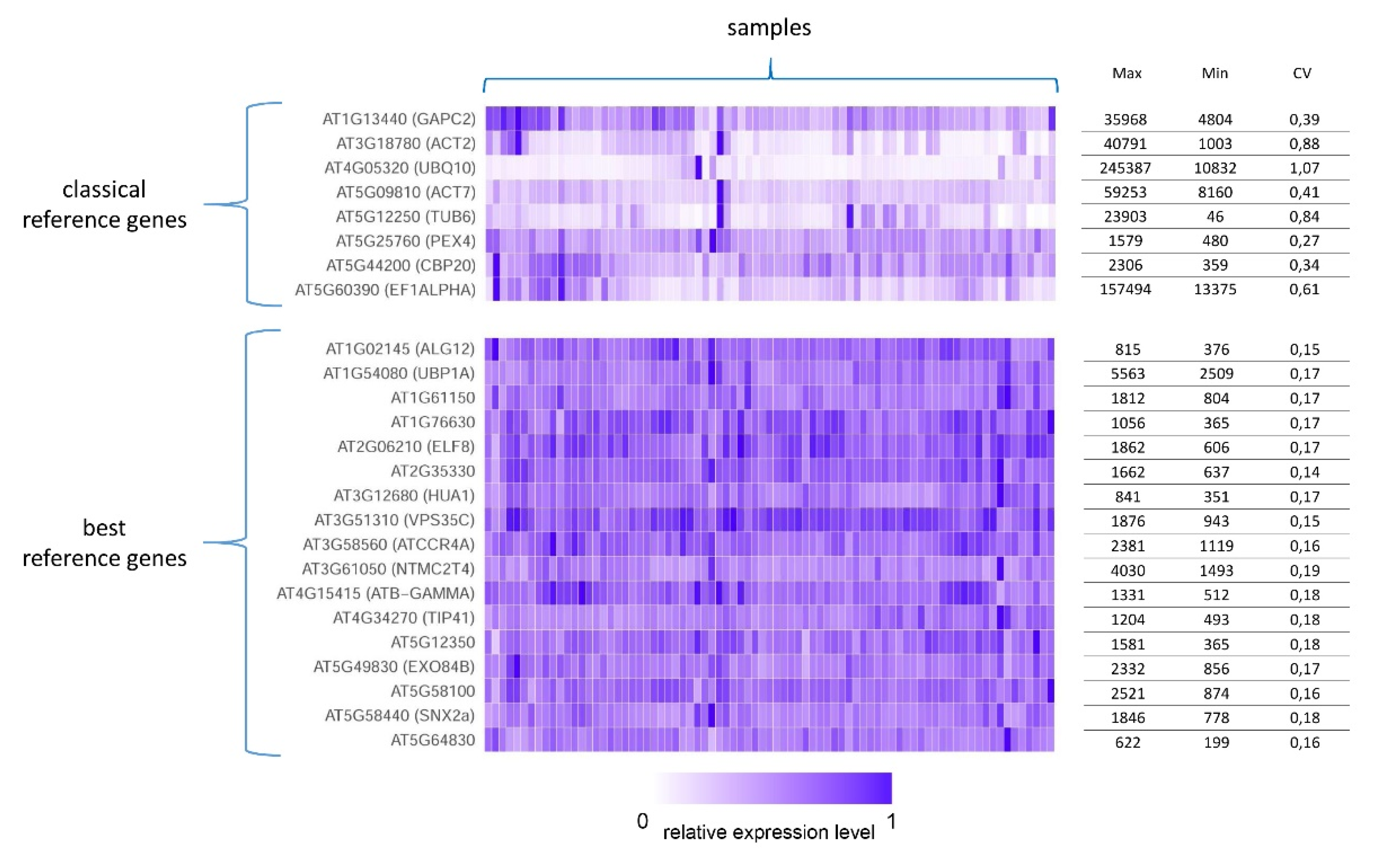
7.1. Stably Expressed Genes
7.2. Tissue-Specific Expression Analysis
7.3. Detailed Studies of Transcription Factor Expression
7.4. Functional Analysis of Genes Using Transcriptome Maps
7.5. Analysis of Evolutionary Processes
7.6. Analysis of Alternative Splicing
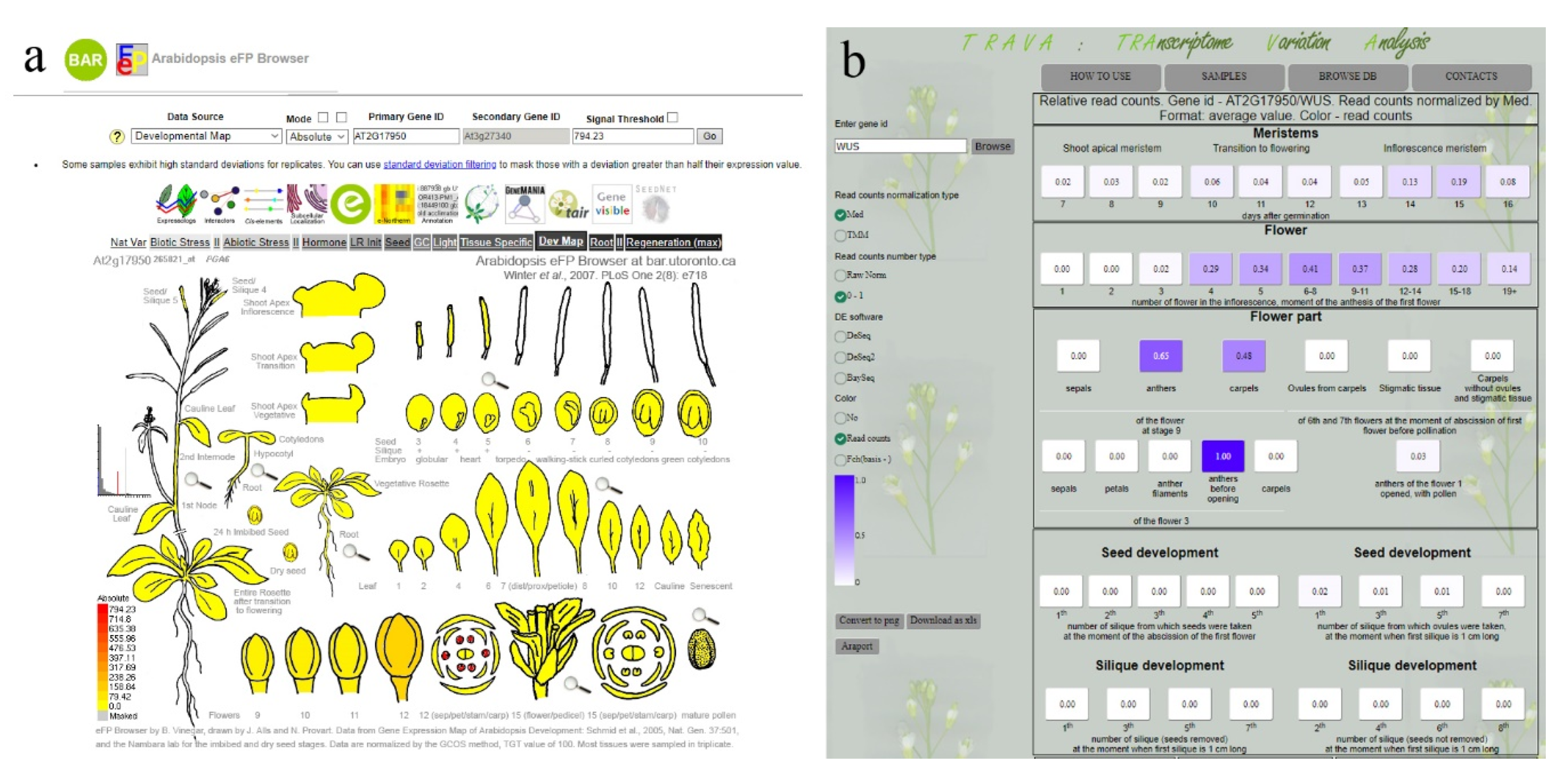
8. How to Summarize the Data–Expression Databases
9. Where to Go Next—Future Perspectives of Transcriptome Maps
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D.; Penin, A.A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016, 88, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Davison, T.S.; Henz, S.R.; Pape, U.J.; Demar, M.; Vingron, M.; Schölkopf, B.; Weigel, D.; Lohmann, J.U. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 2005, 37, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Hanzawa, Y.; Money, T.; Bradley, D. A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. USA 2005, 102, 7748–7753. [Google Scholar] [CrossRef] [PubMed]
- Palovaara, J.; Saiga, S.; Wendrich, J.R.; Hofland, N.V.W.; van Schayck, J.P.; Hater, F.; Mutte, S.; Sjollema, J.; Boekschoten, M.; Hooiveld, G.J.; et al. Transcriptome dynamics revealed by a gene expression atlas of the early Arabidopsis embryo. Nat. Plants 2017, 3, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Meinke, D.W.; Cherry, J.M.; Dean, C.; Rounsley, S.D.; Koornneef, M. Arabidopsis thaliana: A model plant for genome analysis. Science 1998, 282, 662, 679–682. [Google Scholar] [CrossRef]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef]
- Kasianov, A.S.; Klepikova, A.V.; Kulakovskiy, I.V.; Gerasimov, E.S.; Fedotova, A.V.; Besedina, E.G.; Kondrashov, A.S.; Logacheva, M.D.; Penin, A.A. High-quality genome assembly of Capsella bursa-pastoris reveals asymmetry of regulatory elements at early stages of polyploid genome evolution. Plant J. 2017, 91, 278–291. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Debelle, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- The Plant List. The Plant List Version 1.1. Available online: http://www.theplantlist.org/ (accessed on 1 January 2013).
- The Angiosperm Phylogeny Group. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Ortiz-Ramírez, C.; Hernandez-Coronado, M.; Thamm, A.; Catarino, B.; Wang, M.; Dolan, L.; Feijó, J.A.; Becker, J.D. A transcriptome atlas of Physcomitrella patens provides insights into the evolution and development of land plants. Mol. Plant 2016, 9, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Perroud, P.-F.; Haas, F.B.; Hiss, M.; Ullrich, K.K.; Alboresi, A.; Amirebrahimi, M.; Barry, K.; Bassi, R.; Bonhomme, S.; Chen, H.; et al. The Physcomitrella patens gene atlas project: Large-scale RNA-seq based expression data. Plant J. 2018, 95, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Cañas, R.A.; Li, Z.; Pascual, M.B.; Castro-Rodríguez, V.; Ávila, C.; Sterck, L.; Van de Peer, Y.; Cánovas, F.M. The gene expression landscape of pine seedling tissues. Plant J. 2017, 91, 1064–1087. [Google Scholar] [CrossRef]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.-H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef]
- Food and Agricultural Organization of the United Nations. Food and Agricultural Organization of the United Nations Cereal Supply and Demand Brief. Available online: http://www.fao.org/worldfoodsituation/csdb/en/ (accessed on 1 August 2019).
- Schreiber, A.W.; Sutton, T.; Caldo, R.A.; Kalashyan, E.; Lovell, B.; Mayo, G.; Muehlbauer, G.J.; Druka, A.; Waugh, R.; Wise, R.P.; et al. Comparative transcriptomics in the Triticeae. BMC Genom. 2009, 10, 285. [Google Scholar] [CrossRef]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed]
- Nobuta, K.; Venu, R.C.; Lu, C.; Beló, A.; Vemaraju, K.; Kulkarni, K.; Wang, W.; Pillay, M.; Green, P.J.; Wang, G.-l.; et al. An expression atlas of rice mRNAs and small RNAs. Nat. Biotechnol. 2007, 25, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, W.; Chen, Y.; Tang, W.; Yang, J.; Ye, R.; Liu, L.; Lin, Y.; Xu, C.; Xiao, J.; et al. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010, 61, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Lin, H.; Childs, K.L.; Hansey, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Genome-wide atlas of transcription during maize development. Plant J. 2011, 66, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Stelpflug, S.C.; Sekhon, R.S.; Vaillancourt, B.; Hirsch, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Druka, A.; Muehlbauer, G.; Druka, I.; Caldo, R.; Baumann, U.; Rostoks, N.; Schreiber, A.; Wise, R.; Close, T.; Kleinhofs, A.; et al. An atlas of gene expression from seed to seed through barley development. Funct. Integr. Genom. 2006, 6, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Ivamoto, S.T.; Reis, O.; Domingues, D.S.; dos Santos, T.B.; de Oliveira, F.F.; Pot, D.; Leroy, T.; Vieira, L.G.E.; Carazzolle, M.F.; Pereira, G.A.G.; et al. Transcriptome analysis of leaves, flowers and fruits perisperm of Coffea arabica L. reveals the differential expression of genes involved in raffinose biosynthesis. PLoS ONE 2017, 12, e0169595. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.D.; Bombarely, A.; Story, G.W.; Allen, F.; Mueller, L.A.; Coates, S.A.; Jones, L. TobEA: An atlas of tobacco gene expression from seed to senescence. BMC Genom. 2010, 11, 142. [Google Scholar] [CrossRef]
- Massa, A.N.; Childs, K.L.; Lin, H.; Bryan, G.J.; Giuliano, G.; Buell, C.R. The transcriptome of the reference potato genome Solanum tuberosum group Phureja clone DM1-3 516R44. PLoS ONE 2011, 6, e26801. [Google Scholar] [CrossRef]
- Penin, A.A.; Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D. Comparative analysis of developmental transcriptome maps of Arabidopsis thaliana and Solanum lycopersicum. Genes 2019, 10, 50. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [PubMed]
- Sato, S.; Tabata, S.; Hirakawa, H.; Asamizu, E.; Shirasawa, K.; Isobe, S.; Kaneko, T.; Nakamura, Y.; Shibata, D.; Aoki, K.; et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Clevenger, J.; Chu, Y.; Scheffler, B.; Ozias-Akins, P. A developmental transcriptome map for allotetraploid Arachis hypogaea. Front. Plant Sci. 2016, 7, 1446. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Fu, F.; Bucciarelli, B.; Yang, S.S.; Samac, D.A.; Lamb, J.F.S.; Monteros, M.J.; Graham, M.A.; Gronwald, J.W.; Krom, N.; et al. The Medicago sativa gene index 1.2: A web-accessible gene expression atlas for investigating expression differences between Medicago sativa subspecies. BMC Genom. 2015, 16, 502. [Google Scholar] [CrossRef]
- Libault, M.; Farmer, A.; Joshi, T.; Takahashi, K.; Langley, R.J.; Franklin, L.D.; He, J.; Xu, D.; May, G.; Stacey, G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J. 2010, 63, 86–99. [Google Scholar] [CrossRef]
- Severin, A.J.; Woody, J.L.; Bolon, Y.-T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq atlas of glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef]
- Kudapa, H.; Garg, V.; Chitikineni, A.; Varshney, R.K. The RNA-Seq-based high resolution gene expression atlas of chickpea (Cicer arietinum L.) reveals dynamic spatio-temporal changes associated with growth and development. Plant Cell Environ. 2018, 41, 2209–2225. [Google Scholar] [CrossRef]
- Tong, C.; Wang, X.; Yu, J.; Wu, J.; Li, W.; Huang, J.; Dong, C.; Hua, W.; Liu, S. Comprehensive analysis of RNA-seq data reveals the complexity of the transcriptome in Brassica rapa. BMC Genom. 2013, 14, 689. [Google Scholar] [CrossRef]
- Kagale, S.; Nixon, J.; Khedikar, Y.; Pasha, A.; Provart, N.J.; Clarke, W.E.; Bollina, V.; Robinson, S.J.; Coutu, C.; Hegedus, D.D.; et al. The developmental transcriptome atlas of the biofuel crop Camelina sativa. Plant J. 2016, 88, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V. Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol. Biol. Evol. 2003, 20, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Stefanović, S.; Rice, D.W.; Palmer, J.D. Long branch attraction, taxon sampling, and the earliest angiosperms: Amborella or monocots? BMC Evol. Biol. 2004, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Briskine, R.; Hirsch, C.N.; Myers, C.L.; Springer, N.M.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS ONE 2013, 8, e61005. [Google Scholar] [CrossRef] [PubMed]
- Brazma, A.; Hingamp, P.; Quackenbush, J.; Sherlock, G.; Spellman, P.; Stoeckert, C.; Aach, J.; Ansorge, W.; Ball, C.A.; Causton, H.C.; et al. Minimum information about a microarray experiment (MIAME)—Toward standards for microarray data. Nat. Genet. 2001, 29, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Shakoor, N.; Nair, R.; Crasta, O.; Morris, G.; Feltus, A.; Kresovich, S. A Sorghum bicolor expression atlas reveals dynamic genotype-specific expression profiles for vegetative tissues of grain, sweet and bioenergy sorghums. BMC Plant Biol. 2014, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Logan, T.L.; Juenger, T.E. Transcriptome analysis and gene expression atlas for Panicum hallii var. filipes, a diploid model for biofuel research. Plant J. 2012, 70, 879–890. [Google Scholar]
- Vlasova, A.; Capella-Gutiérrez, S.; Rendón-Anaya, M.; Hernández-Oñate, M.; Minoche, A.E.; Erb, I.; Câmara, F.; Prieto-Barja, P.; Corvelo, A.; Sanseverino, W.; et al. Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol. 2016, 17, 32. [Google Scholar] [CrossRef]
- Walley, J.W.; Sartor, R.C.; Shen, Z.; Schmitz, R.J.; Wu, K.J.; Urich, M.A.; Nery, J.R.; Smith, L.G.; Schnable, J.C.; Ecker, J.R.; et al. Integration of omic networks in a developmental atlas of maize. Science 2016, 353, 814–818. [Google Scholar] [CrossRef]
- Dubois, A.; Carrere, S.; Raymond, O.; Pouvreau, B.; Cottret, L.; Roccia, A.; Onesto, J.-P.; Sakr, S.; Atanassova, R.; Baudino, S.; et al. Transcriptome database resource and gene expression atlas for the rose. BMC Genom. 2012, 13, 638. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Bayer, P.E.; Milec, Z.; Vrána, J.; Yuan, Y.; Appels, R.; Edwards, D.; Batley, J.; Nichols, P.; Erskine, W.; et al. An advanced reference genome of Trifolium subterraneum L. reveals genes related to agronomic performance. Plant Biotechnol. J. 2017, 15, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Tausta, S.L.; Gandotra, N.; Sun, N.; Liu, T.; Clay, N.K.; Ceserani, T.; Chen, M.; Ma, L.; Holford, M.; et al. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat. Genet. 2009, 41, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.-L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-lengthde novoassembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Farmer, A.; Brechenmacher, L.; Drnevich, J.; Langley, R.J.; Bilgin, D.D.; Radwan, O.; Neece, D.J.; Clough, S.J.; May, G.D.; et al. Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 2010, 152, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Czechowski, T. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Ubi, B.E.; Saito, T.; Moriguchi, T. Evaluation of reference genes for accurate normalization of gene expression for real time-quantitative PCR in Pyrus pyrifolia using different tissue samples and seasonal conditions. PLoS ONE 2014, 9, e86492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Q.; Sun, M.; Zhu, L.; Yang, M.; Zhao, Y. Selection of reference genes for quantitative real-time PCR normalization in Panax ginseng at different stages of growth and in different organs. PLoS ONE 2014, 9, e112177. [Google Scholar] [CrossRef]
- Niu, J.; Zhu, B.; Cai, J.; Li, P.; Wang, L.; Dai, H.; Qiu, L.; Yu, H.; Ha, D.; Zhao, H.; et al. Selection of reference genes for gene expression studies in Siberian Apricot (Prunus sibirica L.) germplasm using quantitative real-time PCR. PLoS ONE 2014, 9, e103900. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Blumwald, E. Assessing reference genes for accurate transcript normalization using quantitative real-time PCR in Pearl Millet [Pennisetum glaucum (L.) R. Br.]. PLoS ONE 2014, 9, e106308. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; He, B.; Gao, X.; Qin, Y.; Yang, J.; Fang, Y.; Qi, J.; Tang, C. Validation of reference genes for quantitative real-time PCR during latex regeneration in rubber tree. Gene 2015, 563, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Kozera, B.; Rapacz, M. Reference genes in real-time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Torres-Jerez, I.; Wang, M.; Andriankaja, A.; Allen, S.N.; He, J.; Tang, Y.; Murray, J.D.; Udvardi, M.K. Establishment of the Lotus japonicus gene expression atlas (LjGEA) and its use to explore legume seed maturation. Plant J. 2013, 74, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Jiang, C.; Huang, Z.; Torres-Jerez, I.; Chang, J.; Zhang, H.; Udvardi, M.; Liu, R.; Verdier, J. The Vigna unguiculata gene expression atlas (VuGEA) from de novo assembly and quantification of RNA-seq data provides insights into seed maturation mechanisms. Plant J. 2016, 88, 318–327. [Google Scholar] [CrossRef]
- Quesada, T.; Li, Z.; Dervinis, C.; Li, Y.; Bocock, P.N.; Tuskan, G.A.; Casella, G.; Davis, J.M.; Kirst, M. Comparative analysis of the transcriptomes of Populus trichocarpa and Arabidopsis thaliana suggests extensive evolution of gene expression regulation in angiosperms. New Phytol. 2008, 180, 408–420. [Google Scholar] [CrossRef]
- Xiao, S.J.; Zhang, C.; Zou, Q.; Ji, Z.L. TiSGeD: A database for tissue-specific genes. Bioinformatics 2010, 26, 1273–1275. [Google Scholar] [CrossRef]
- Sibout, R.; Proost, S.; Hansen, B.O.; Vaid, N.; Giorgi, F.M.; Ho-Yue-Kuang, S.; Legée, F.; Cézart, L.; Bouchabké-Coussa, O.; Soulhat, C.; et al. Expression atlas and comparative coexpression network analyses reveal important genes involved in the formation of lignified cell wall in Brachypodium distachyon. New Phytol. 2017, 215, 1009–1025. [Google Scholar] [CrossRef]
- Schug, J.; Schuller, W.-P.; Kappen, C.; Salbaum, J.M.; Bucan, M.; Stoeckert, C.J. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005, 6, R33. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lin, Y.; Nery, J.R.; Urich, M.A.; Breschi, A.; Davis, C.A.; Dobin, A.; Zaleski, C.; Beer, M.A.; Chapman, W.C.; et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 17224–17229. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [PubMed]
- Fasoli, M.; Dal Santo, S.; Zenoni, S.; Tornielli, G.B.; Farina, L.; Zamboni, A.; Porceddu, A.; Venturini, L.; Bicego, M.; Murino, V.; et al. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. The Plant Cell 2012, 24, 3489–3505. [Google Scholar] [CrossRef] [PubMed]
- Høgslund, N.; Radutoiu, S.; Krusell, L.; Voroshilova, V.; Hannah, M.A.; Goffard, N.; Sanchez, D.H.; Lippold, F.; Ott, T.; Sato, S.; et al. Dissection of symbiosis and organ development by integrated transcriptome analysis of Lotus japonicus mutant and wild-type plants. PLoS ONE 2009, 4, e6556. [Google Scholar] [CrossRef]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed]
- Waese, J.; Fan, J.; Pasha, A.; Yu, H.; Fucile, G.; Shi, R.; Cumming, M.; Kelley, L.A.; Sternberg, M.J.; Krishnakumar, V.; et al. ePlant: Visualizing and exploring multiple levels of data for hypothesis generation in plant biology. Plant Cell 2017, 29, 1806–1821. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Tamiru, M.; Salby, R.; Bolitho, C.; Williams, A.; Huggard, S.; Fisch, E.; Unsworth, K.; Whelan, J.; Lewsey, M.G. AgriSeqDB: An online RNA-Seq database for functional studies of agriculturally relevant plant species. BMC Plant Biol. 2018, 19, 200. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Nakamura, Y.; Kobayashi, M.; Terashima, S.; Ishizuka, W.; Uchiyama, K.; Tsumura, Y.; Yano, K.; Goto, S. TodoFirGene: Developing Transcriptome Resources for Genetic Analysis of Abies sachalinensis. Plant Cell Physiol. 2018, 59, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Yano, R.; Nonaka, S.; Ezura, H. Melonet-DB, a grand RNA-Seq gene expression atlas in melon (Cucumis melo L.). Plant Cell Physiol. 2018, 59, e4. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Benedito, V.A.; Wang, M.; Murray, J.D.; Zhao, P.X.; Tang, Y.; Udvardi, M.K. The Medicago truncatula gene expression atlas web server. BMC Bioinform. 2009, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- Zouine, M.; Maza, E.; Djari, A.; Lauvernier, M.; Frasse, P.; Smouni, A.; Pirrello, J.; Bouzayen, M. TomExpress, a unified tomato RNA-Seq platform for visualization of expression data, clustering and correlation networks. Plant J. 2017, 92, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Ding, X.B.; Li, X.; Jin, C.F.; Yue, Y.W.; Li, G.P.; Guo, H. An atlas and analysis of bovine skeletal muscle long noncoding RNAs. Anim. Genet. 2017, 48, 278–286. [Google Scholar] [CrossRef]
- Davie, K.; Janssens, J.; Koldere, D.; De Waegeneer, M.; Pech, U.; Kreft, Ł.; Aibar, S.; Makhzami, S.; Christiaens, V.; González-Blas, C.B.; et al. A single-cell transcriptome atlas of the aging Drosophila brain. Cell 2018, 174, 982–998.e20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Deng, F.; Jia, X.; Li, C.; Lai, S.-J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep. 2017, 7, 7648. [Google Scholar] [CrossRef] [PubMed]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M.; et al. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 2019, 27, 2241–2247. [Google Scholar] [CrossRef]
- Giacomello, S.; Salmén, F.; Terebieniec, B.K.; Vickovic, S.; Navarro, J.F.; Alexeyenko, A.; Reimegård, J.; McKee, L.S.; Mannapperuma, C.; Bulone, V.; et al. Spatially resolved transcriptome profiling in model plant species. Nat. Plants. 2017, 8, 17061. [Google Scholar] [CrossRef]
- Giacomello, S.; Lundeberg, J. Preparation of plant tissue to enable Spatial Transcriptomics profiling using barcoded microarrays. Nat. Protoc. 2018, 13, 2425–2446. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klepikova, A.V.; Penin, A.A. Gene Expression Maps in Plants: Current State and Prospects. Plants 2019, 8, 309. https://doi.org/10.3390/plants8090309
Klepikova AV, Penin AA. Gene Expression Maps in Plants: Current State and Prospects. Plants. 2019; 8(9):309. https://doi.org/10.3390/plants8090309
Chicago/Turabian StyleKlepikova, Anna V., and Aleksey A. Penin. 2019. "Gene Expression Maps in Plants: Current State and Prospects" Plants 8, no. 9: 309. https://doi.org/10.3390/plants8090309
APA StyleKlepikova, A. V., & Penin, A. A. (2019). Gene Expression Maps in Plants: Current State and Prospects. Plants, 8(9), 309. https://doi.org/10.3390/plants8090309