Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome


Abstract
1. Introduction
2. Results and Discussion
2.1. Analysis and Discussion
2.1.1. Characters of Rhodomyrtus tomentosa Chloroplast (CP) Genome
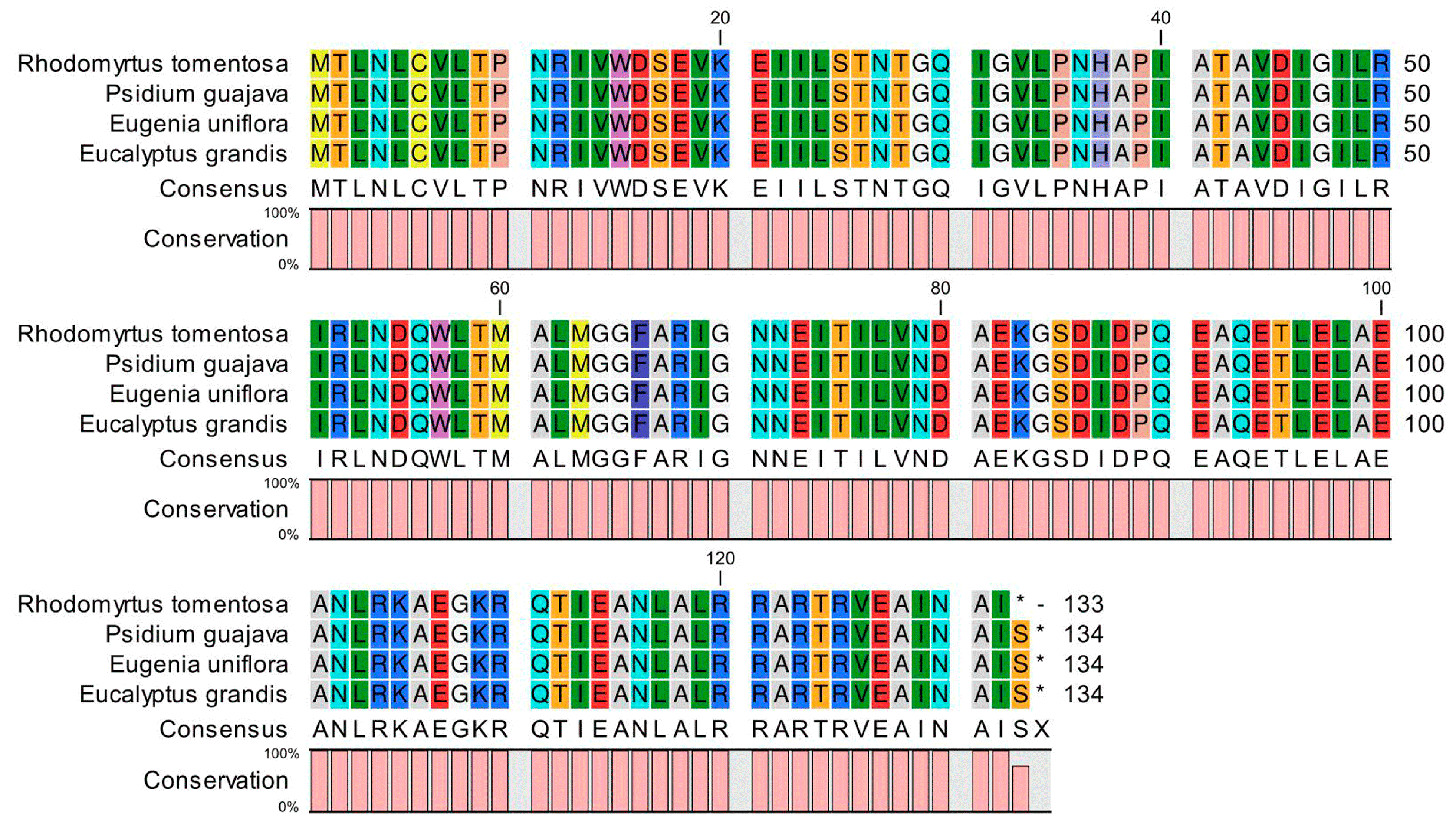
2.1.2. Analysis of Premature Termination Codons in the R. tomentosa CP Genome
2.1.3. Identification of Long Repeats (LRs) and Simple Sequence Repeats (SSRs)
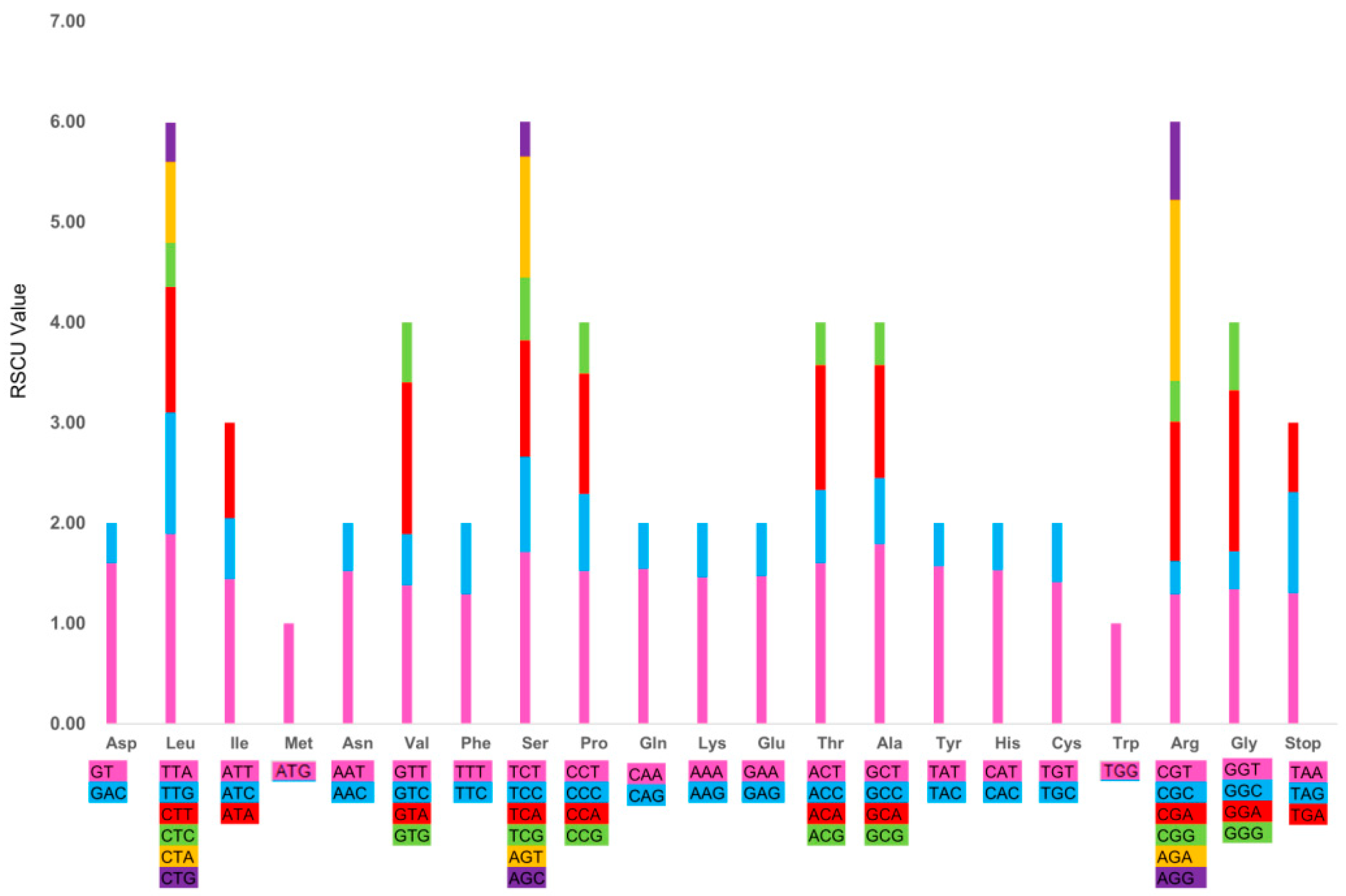
2.1.4. Codon Usage and RNA Editing Sites
2.1.5. Contraction and Expansion of IRs in the R. tomentosa CP Genome
2.1.6. Comparative CP Genomic Analysis
2.1.7. Phylogenetic Analysis of the R. tomentosa CP Genome
3. Materials and Methods
3.1. Plant Material, DNA Extraction and Sequencing
3.2. Chloroplast Genome Assembly and Annotation
3.3. Genome Structure and Genome Comparison
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freire, C.G.; Giachini, A.J.; Gardin, J.P.P.; Rodrigues, A.C.; Vieira, R.L.; Baratto, C.M.; Werner, S.S.; Abreu, B.H. First record of in vitro formation of ectomycorrhizae in Psidium cattleianum Sabine, a native Myrtaceae of the Brazilian Atlantic Forest. PLoS ONE 2018, 13, e0196984. [Google Scholar] [CrossRef] [PubMed]
- He, S.M.; Wang, X.; Yang, S.C.; Dong, Y.; Zhao, Q.M.; Yang, J.L.; Cong, K.; Zhang, J.J.; Zhang, G.H.; Wang, Y.; et al. De novo Transcriptome Characterization of Rhodomyrtus tomentosa Leaves and Identification of Genes Involved in alpha/beta-Pinene and beta-Caryophyllene Biosynthesis. Front. Plant Sci. 2018, 9, 1231. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Ma, G.; Li, N.; Deng, Q.; Yin, Y.; Huang, R. Investigation of in vitro and in vivo antioxidant activities of flavonoids rich extract from the berries of Rhodomyrtus tomentosa(Ait.) Hassk. Food Chem. 2015, 173, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, J.G.; Su, J.C.; Huang, X.J.; Ye, W.C.; Wang, Y. Tomentodione E, a new sec-pentyl syncarpic acid-based meroterpenoid from the leaves of Rhodomyrtus tomentosa. J. Asian Nat. Prod. Res. 2018, 20, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Chen, C.; Wang, X.B.; Wu, L.; Yang, M.H.; Luo, J.; Zhang, C.; Sun, H.B.; Luo, J.G.; Kong, L.Y. Rhodomyrtials A and B, Two Meroterpenoids with a Triketone-Sesquiterpene-Triketone Skeleton from Rhodomyrtus tomentosa: Structural Elucidation and Biomimetic Synthesis. Org. Lett. 2016, 18, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, G.; Voravuthikunchai, S.P.; Towatana, N.H. Acetone Extract from Rhodomyrtus tomentosa: A Potent Natural Antioxidant. Evid. Based Complement. Altern. Med. 2012, 2012, 535479. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Kaila, T.; Chaduvla, P.K.; Rawal, H.C.; Saxena, S.; Tyagi, A.; Mithra, S.V.A.; Solanke, A.U.; Kalia, P.; Sharma, T.R.; Singh, N.K.; et al. Chloroplast Genome Sequence of Clusterbean (Cyamopsis tetragonoloba L.): Genome Structure and Comparative Analysis. Genes 2017, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Cremen, M.C.M.; Leliaert, F.; West, J.; Lam, D.W.; Shimada, S.; Lopez-Bautista, J.M.; Verbruggen, H. Reassessment of the classification of Bryopsidales (Chlorophyta) based on chloroplast phylogenomic analyses. Mol. Phylogenetics Evol. 2019, 130, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Tabata, S. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 1999, 6, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, D.; Song, G.; Wei, X.; Chen, L.; Wu, X.; Li, X.; Zhu, Z. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci. China Ser. Life Sci. 2003, 46, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, K. Chloroplast and mitochondrial genomes from a liverwort, Marchantia polymorpha--gene organization and molecular evolution. Biosci. Biotechnol. Biochem. 1996, 60, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shao, J.; Zhang, H.; Jiang, M.; Huang, L.; Zhang, Z.; Yang, D.; He, M.; Ronaghi, M.; Luo, X.; et al. Sequencing and Analysis of Strobilanthes cusia (Nees) Kuntze Chloroplast Genome Revealed the Rare Simultaneous Contraction and Expansion of the Inverted Repeat Region in Angiosperm. Front. Plant Sci. 2018, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Kwak, M.; Lee, B.; Park, S. Complete chloroplast genome of Tetragonia tetragonioides: Molecular phylogenetic relationships and evolution in Caryophyllales. PLoS ONE 2018, 13, e0199626. [Google Scholar] [CrossRef]
- Trosch, R.; Barahimipour, R.; Gao, Y.; Badillo-Corona, J.A.; Gotsmann, V.L.; Zimmer, D.; Muhlhaus, T.; Zoschke, R.; Willmund, F. Commonalities and differences of chloroplast translation in a green alga and land plants. Nat. plants 2018, 4, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Paiva, J.A.; Prat, E.; Vautrin, S.; Santos, M.D.; San-Clemente, H.; Brommonschenkel, S.; Fonseca, P.G.; Grattapaglia, D.; Song, X.; Ammiraju, J.S.; et al. Advancing Eucalyptus genomics: Identification and sequencing of lignin biosynthesis genes from deep-coverage BAC libraries. BMC Genom. 2011, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.; Schairer, H.U.; Sebald, W. Identification of amino-acid substitutions in the proteolipid subunit of the ATP synthase from dicyclohexylcarbodiimide-resistant mutants of Escherichia coli. Eur. J. Biochem. 1980, 112, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bukowy-Bieryllo, Z.; Dabrowski, M.; Witt, M.; Zietkiewicz, E. Aminoglycoside-stimulated readthrough of premature termination codons in selected genes involved in primary ciliary dyskinesia. RNA Biol. 2016, 13, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, H.; Wang, L.; Zhu, C.; Sheng, K.; Du, Y.; Wang, K.; Dias, A.; Chen, S.; Whitman, M.; et al. Premature Termination Codons Are Recognized in the Nucleus in A Reading-Frame Dependent Manner. Cell Discov. 2015, 1, 15001. [Google Scholar] [CrossRef]
- Sui, T.; Song, Y.; Liu, Z.; Chen, M.; Deng, J.; Xu, Y.; Lai, L.; Li, Z. CRISPR-induced exon skipping is dependent on premature termination codon mutations. Genome Biol. 2018, 19, 164. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of the Medicinal Plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.H.; Jiang, X.M.; Yan, C.C.; Zhang, W.Q.; Xiao, G.S.; Yue, B.S.; Zhou, C.Q. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 2018, 8, 14407. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Shanker, A. Identification of Simple Sequence Repeats in Chloroplast Genomes of Magnoliids Through Bioinformatics Approach. Interdiscip. Sci. 2016, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y.; Song, J.; Yao, H. Molecular Structure and Phylogenetic Analyses of Complete Chloroplast Genomes of Two Aristolochia Medicinal Species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Taylor, D.R. Testing for selection on synonymous sites in plant mitochondrial DNA: The role of codon bias and RNA editing. J. Mol. Evol. 2010, 70, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Complete Chloroplast Genome Sequence of Decaisnea insignis: Genome Organization, Genomic Resources and Comparative Analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Luo, C. Molecular and Functional Diversity of RNA Editing in Plant Mitochondria. Mol. Biotechnol. 2018, 60, 935–945. [Google Scholar] [CrossRef]
- Reginato, M.; Neubig, K.M.; Majure, L.C.; Michelangeli, F.A. The first complete plastid genomes of Melastomataceae are highly structurally conserved. Peer J. 2016, 4, e2715. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhihai, H.; Jiang, X.; Shuiming, X.; Baosheng, L.; Yuan, G.; Chaochao, Z.; Xiaohui, Q.; Wen, X.; Shilin, C. Comparative optical genome analysis of two pangolin species: Manis pentadactyla and Manis javanica. GigaScience 2016, 5, 1–5. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Positions | T (%) | C (%) | A (%) | G (%) | Length (bp) |
|---|---|---|---|---|---|---|
| LSC | 33.3 | 18.0 | 31.7 | 17.1 | 86,298 | |
| IRA | 28.4 | 20.7 | 28.7 | 22.2 | 25,824 | |
| SSC | 34.1 | 16.2 | 35.1 | 14.6 | 18,183 | |
| IRB | 28.7 | 22.2 | 28.4 | 20.7 | 25,284 | |
| Total | 31.8 | 18.9 | 31.0 | 18.2 | 156,129 | |
| CDS | 31.6 | 17.5 | 30.8 | 20.1 | 76,113 | |
| 1st position | 24.0 | 18.6 | 30.8 | 26.3 | 25,371 | |
| 2nd position | 33.0 | 19.8 | 29.6 | 17.5 | 25,371 | |
| 3rd position | 37.0 | 14.1 | 32.0 | 16.5 | 25,371 |
| Gene Classification | Gene Names | Number |
|---|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 |
| Cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 |
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | 6 |
| NADH dehydrogenase | ndhA *, ndhB * (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12(1) |
| RuBisCO large subunit | rbcL | 1 |
| RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | 4 |
| Ribosomal proteins (SSC) | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 ** (×2), rps14, rps15, rps16 *, rps18, rps19 | 14(2) |
| Ribosomal proteins (LSC) | rpl2 (×2), rpl14, rpl16, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 | 11 |
| Ribosomal RNAs | rrn 4.5 (×2), rrn 5 (×2), rrn 16 (×2), rrn 23 (×2) | 8(4) |
| Protein of unknown function | ycf1, ycf2 (×2), ycf3 **, ycf4 | 5(1) |
| Transfer RNAs | 37 tRNAs (8 contain an intron, 7 in the inverted repeats region) | 37(7) |
| Other genes | accD, ccsA, cemA, clpP, matK | 5 |
| Total | 129 |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| atpF | LSC | 144 | 739 | 411 | ||
| clpP | LSC | 69 | 852 | 296 | 637 | 228 |
| ndhA | SSC | 552 | 1058 | 540 | ||
| ndhB | IR | 777 | 681 | 756 | ||
| petB | LSC | 6 | 778 | 642 | ||
| petD | LSC | 7 | 750 | 473 | ||
| rpl16 | LSC | 9 | 988 | 399 | ||
| rpl2 | IR | 391 | 664 | 434 | ||
| rpoC1 | LSC | 451 | 733 | 1619 | ||
| rps12 | LSC | 114 | 232 | 546 | 26 | |
| rps16 | LSC | 40 | 860 | 212 | ||
| trnA-UGC | IR | 35 | 803 | 38 | ||
| trnG-UCC | LSC | 23 | 734 | 48 | ||
| trnI-GAU | IR | 37 | 949 | 35 | ||
| trnK-UUU | LSC | 35 | 2469 | 37 | ||
| trnL-UAA | LSC | 35 | 505 | 50 | ||
| trnV-UAC | LSC | 37 | 587 | 37 | ||
| ycf3 | LSC | 126 | 754 | 226 | 723 | 155 |
| SSR Type | Repeat Unit | Amount | Ratio (%) |
|---|---|---|---|
| Mono | A/T | 171 | 98.8 |
| C/G | 2 | 1.2 | |
| Di | AC/GT | 16 | 43.2 |
| AT/AT | 21 | 56.8 | |
| Tri | AAC/GTT | 6 | 9.5 |
| AAG/CTT | 18 | 28.6 | |
| AAT/ATT | 24 | 38.1 | |
| ACC/GGT | 2 | 3.2 | |
| ACT/AGT | 3 | 4.8 | |
| AGC/CTG | 5 | 7.9 | |
| AGG/CCT | 1 | 1.6 | |
| ATC/ATG | 4 | 6.3 | |
| Tetra | AAAG/CTTT | 2 | 16.7 |
| AAAT/ATTT | 3 | 25.0 | |
| AAGC/CTTG | 1 | 8.3 | |
| AAGT/ACTT | 1 | 8.3 | |
| AATT/AATT | 2 | 16.7 | |
| AGAT/ATCT | 3 | 25.0 | |
| Penta | ACCGG/CCGGT | 2 | 100.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Yang, Z.; Huang, S.; An, W.; Li, J.; Zheng, X. Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome. Plants 2019, 8, 89. https://doi.org/10.3390/plants8040089
Huang Y, Yang Z, Huang S, An W, Li J, Zheng X. Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome. Plants. 2019; 8(4):89. https://doi.org/10.3390/plants8040089
Chicago/Turabian StyleHuang, Yuying, Zerui Yang, Song Huang, Wenli An, Jing Li, and Xiasheng Zheng. 2019. "Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome" Plants 8, no. 4: 89. https://doi.org/10.3390/plants8040089
APA StyleHuang, Y., Yang, Z., Huang, S., An, W., Li, J., & Zheng, X. (2019). Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome. Plants, 8(4), 89. https://doi.org/10.3390/plants8040089