Isoform-Specific NO Synthesis by Arabidopsis thaliana Nitrate Reductase

Abstract
1. Introduction
2. Results
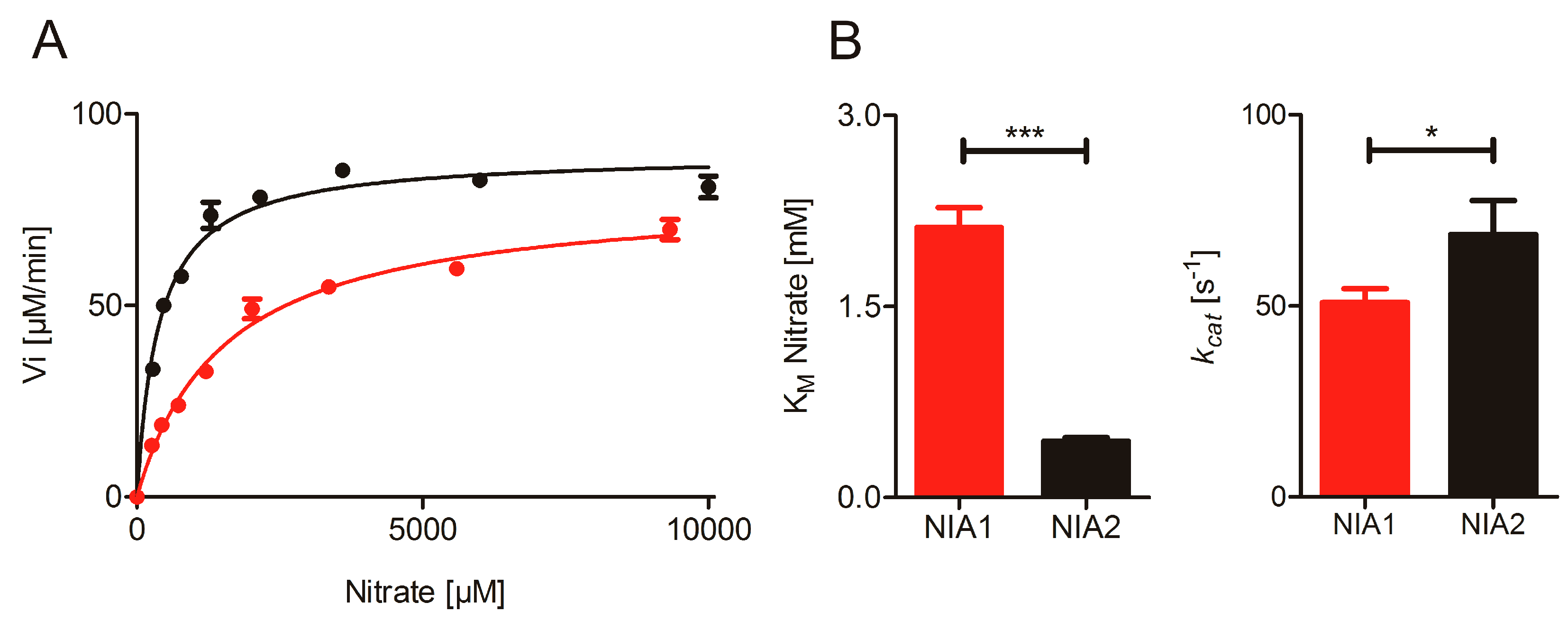
2.1. Nitrate Reduction Activity
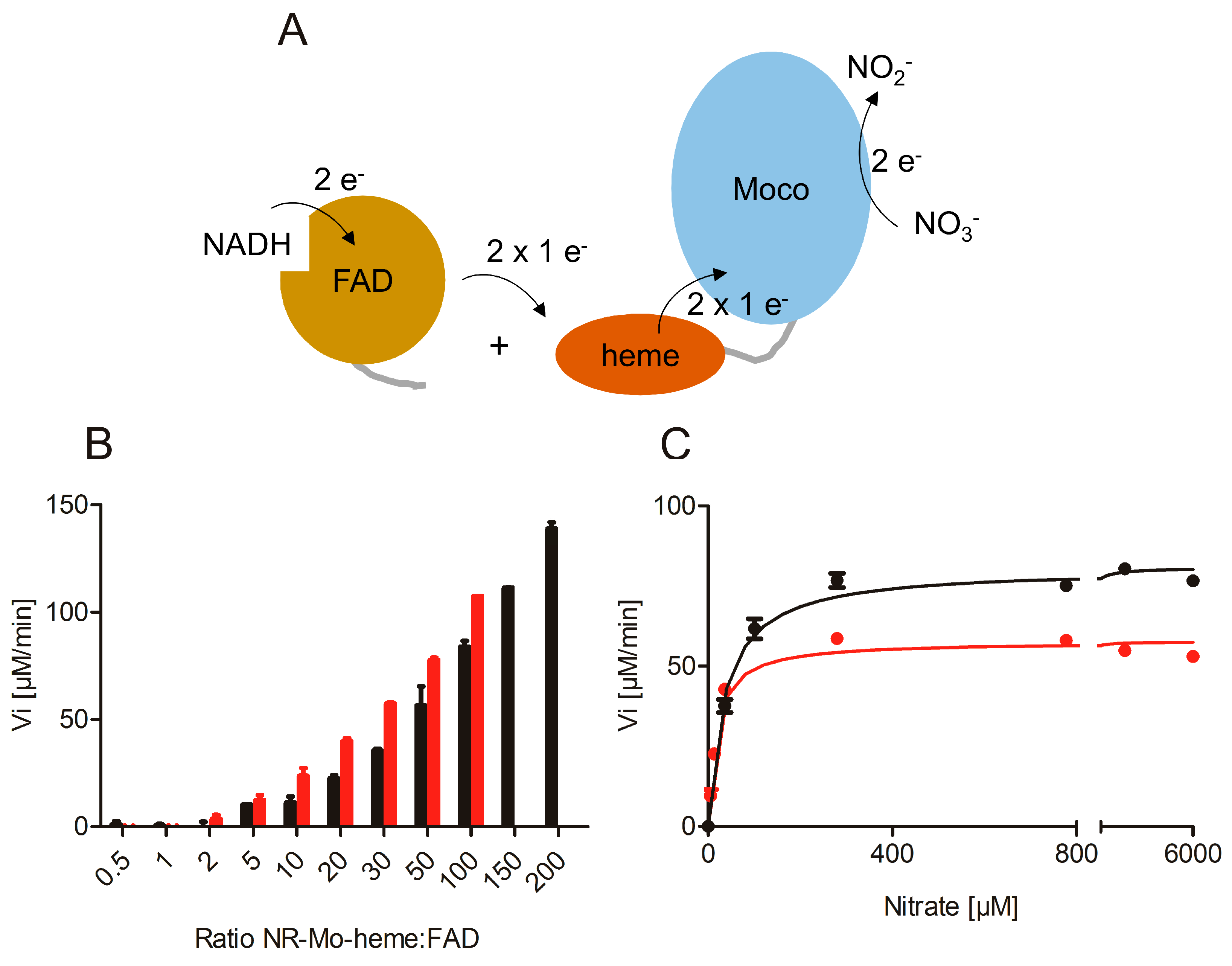
2.2. Re-Constitution of Full-Length NR activity In Vitro
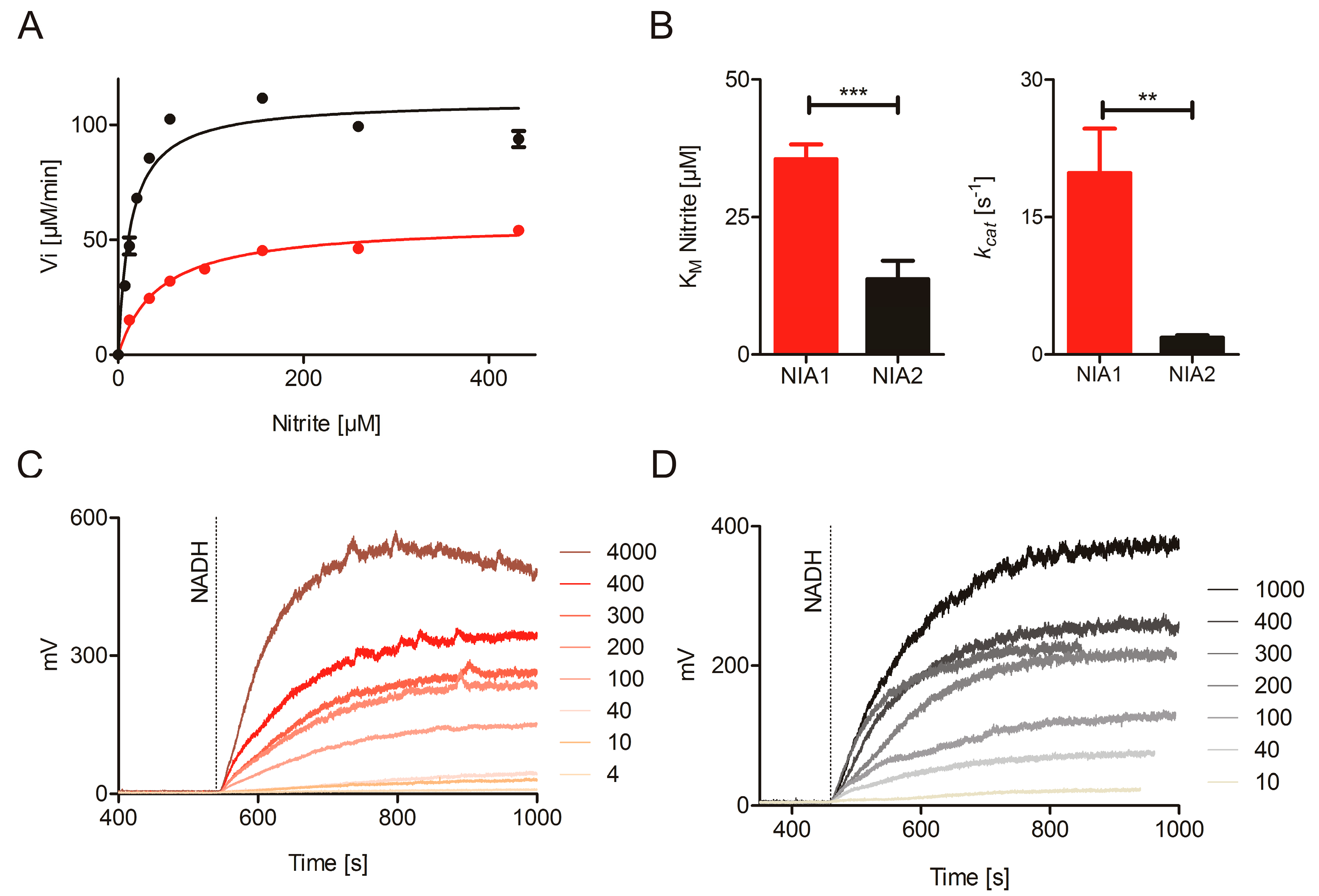
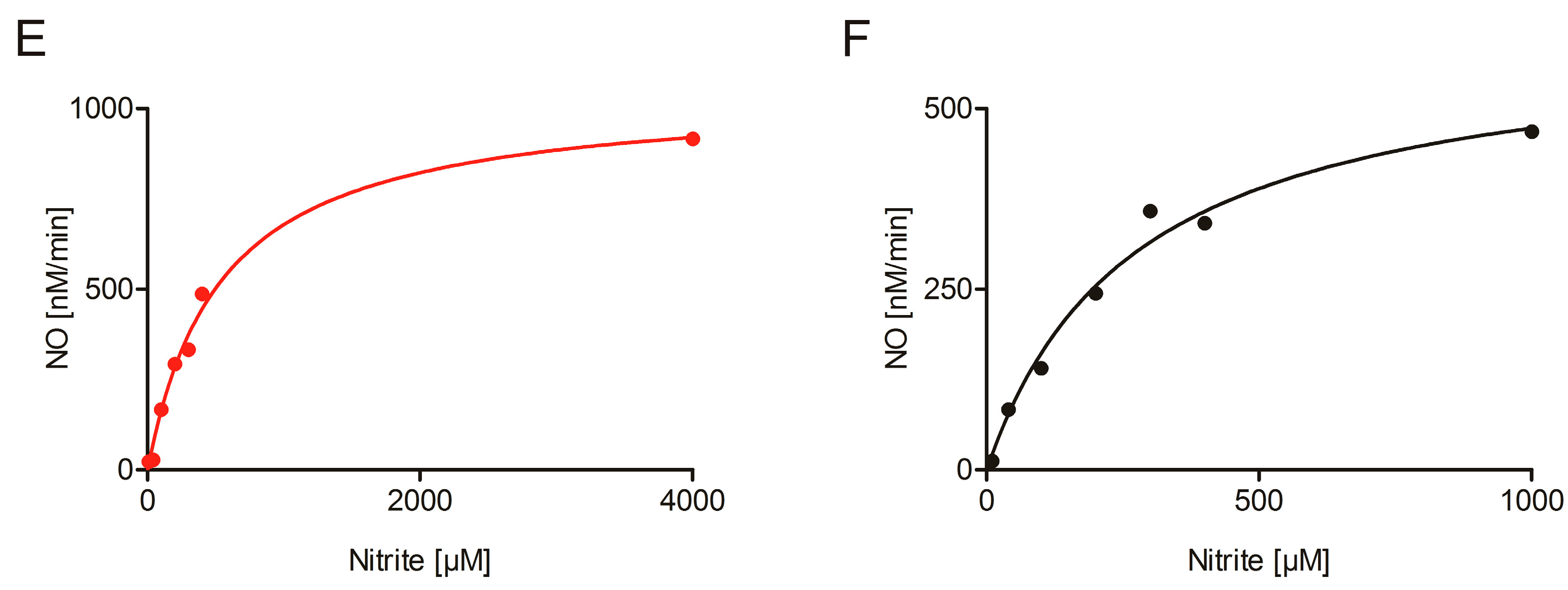
2.3. Nitrite Reduction Activity
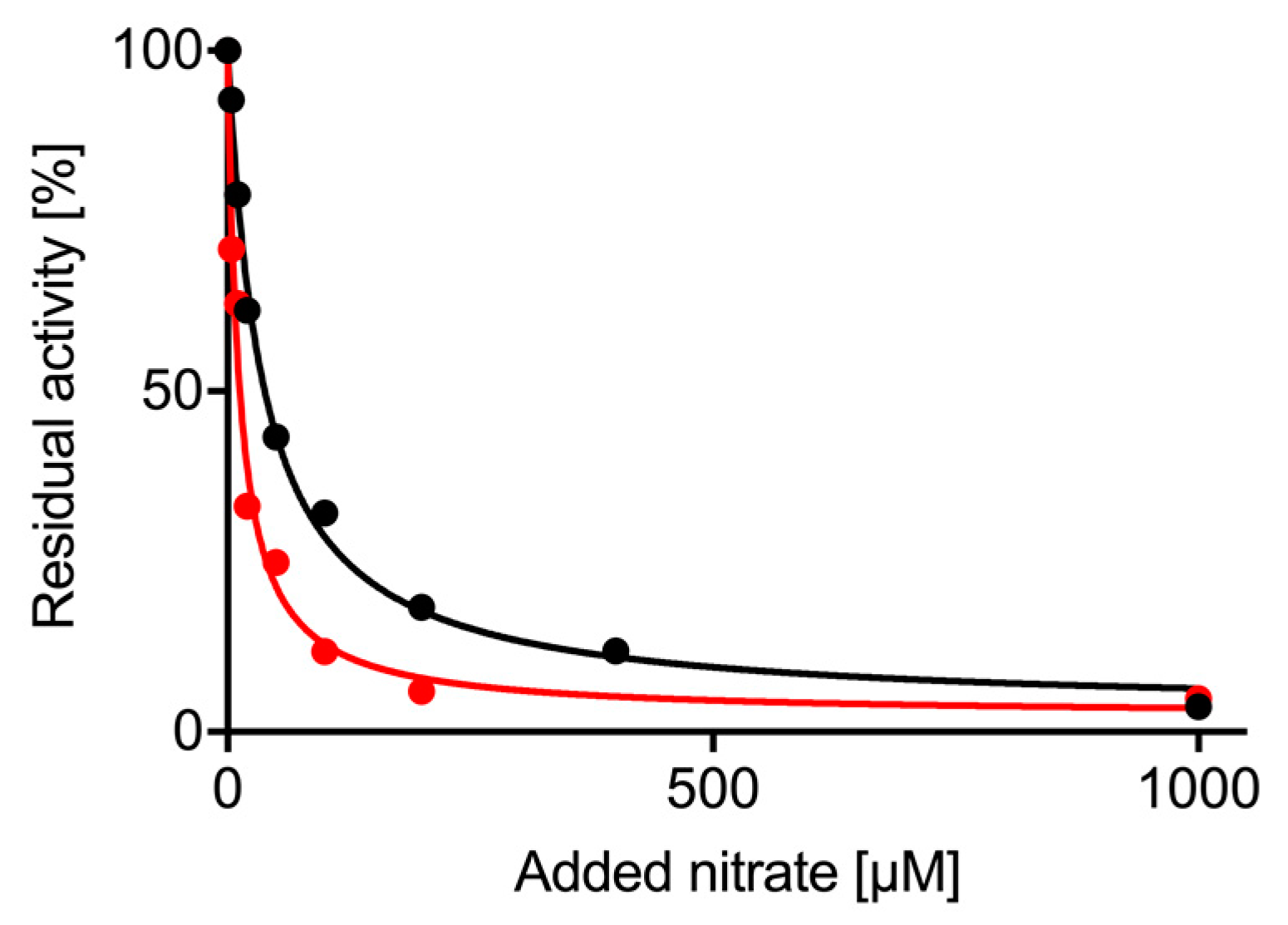
2.4. Nitrate Inhibition of Nitrite Reductase Activity
3. Discussion
4. Materials and Methods
4.1. Recombinant Proteins
4.2. SDS-PAGE and Western Blot
4.3. Enyzme Assays
4.4. NO Quantification Using the NO-Analyzer
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, W.H. Nitrate reductase and its role in nitrate assimilation in plants. Physiol. Plantarum. 1988, 74, 214–219. [Google Scholar] [CrossRef]
- Meyer, C.; Stitt, M. Nitrate reduction and signalling. In Plant Nitrogen; Lea, P.J., Morot-Gaudry, J.-F., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2001; pp. 37–59. [Google Scholar]
- Bachmann, M.; Shiraishi, N.; Campbell, W.H.; Yoo, B.C.; Harmon, A.C.; Huber, S.C. Identification of ser-543 as the major regulatory phosphorylation site in spinach leaf nitrate reductase. Plant Cell 1996, 8, 505–517. [Google Scholar] [CrossRef]
- Su, W.; Huber, S.C.; Crawford, N.M. Identification in vitro of a post-translational regulatory site in the hinge 1 region of arabidopsis nitrate reductase. Plant Cell 1996, 8, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lambeck, I.C.; Fischer-Schrader, K.; Niks, D.; Roeper, J.; Chi, J.C.; Hille, R.; Schwarz, G. Molecular mechanism of 14-3-3 protein-mediated inhibition of plant nitrate reductase. J. Bio. Chem. 2012, 287, 4562–4571. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.C.; Roeper, J.; Schwarz, G.; Fischer-Schrader, K. Dual binding of 14-3-3 protein regulates arabidopsis nitrate reductase activity. J. Biol. Inorg. Chem. 2015, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.E. Evolution of nitrogen oxide(s) during in vivo nitrate reductase assay of soybean leaves. Plant Physiol. 1981, 68, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Wildt, J.; Kley, D.; Rockel, A.; Rockel, P.; Segschneider, H.J. Emission of no from several higher plant species. J. Geophys. Res. 1997, 102, 5919–5927. [Google Scholar] [CrossRef]
- Yamasaki, H.; Sakihama, Y. Simultaneous production of nitric oxide and peroxynitrite by plant nitrate reductase: In vitro evidence for the NR-dependent formation of active nitrogen species. FEBS Lett. 2000, 468, 89–92. [Google Scholar] [CrossRef]
- Wendehenne, D.; Hancock, J.T. New frontiers in nitric oxide biology in plant. Plant Sci. Int. J. Exp. Plant Biol. 2011, 181, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Farnese, F.S.; Menezes-Silva, P.E.; Gusman, G.S.; Oliveira, J.A. When bad guys become good ones: The key role of reactive oxygen species and nitric oxide in the plant response to abiotic stress. Front. Plant Sci. 2016, 7, 471. [Google Scholar] [CrossRef]
- Sanz-Luque, E.; Chamizo-Ampudia, A.; Llamas, A.; Galvan, A.; Fernandez, E. Understanding nitrate assimilation and its regulation in microalgae. Front. Plant Sci. 2015, 6, 899. [Google Scholar] [CrossRef] [PubMed]
- Bethke, P.C.; Badger, M.R.; Jones, R.L. Apoplastic synthesis of nitric oxide by plant tissues. Plant Cell 2004, 16, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Barroso, J.B.; Corpas, F.J.; Carreras, A.; Sandalio, L.M.; Valderrama, R.; Palma, J.M.; Lupianez, J.A.; del Rio, L.A. Localization of nitric-oxide synthase in plant peroxisomes. J. Biol. Chem. 1999, 274, 36729–36733. [Google Scholar] [CrossRef]
- Fröhlich, A.; Durner, J. The hunt for plant nitric oxide synthase (NOS): Is one really needed? Plant Sci. 2011, 181, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Alber, N.A.; Sivanesan, H.; Vanlerberghe, G.C. The occurrence and control of nitric oxide generation by the plant mitochondrial electron transport chain. Plant Cell Environ. 2017, 40, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.; Schwarz, G. Nitrite-dependent nitric oxide synthesis by molybdenum enzymes. FEBS Lett. 2018, 592, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.J.; Fernie, A.R.; Kaiser, W.M.; van Dongen, J.T. On the origins of nitric oxide. Trends Plant Sci. 2011, 16, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Mur, L.A.; Mandon, J.; Persijn, S.; Cristescu, S.M.; Moshkov, I.E.; Novikova, G.V.; Hall, M.A.; Harren, F.J.; Hebelstrup, K.H.; Gupta, K.J. Nitric oxide in plants: An assessment of the current state of knowledge. AoB Plants 2013, 5, pls052. [Google Scholar] [CrossRef] [PubMed]
- Planchet, E.; Jagadis Gupta, K.; Sonoda, M.; Kaiser, W.M. Nitric oxide emission from tobacco leaves and cell suspensions: Rate limiting factors and evidence for the involvement of mitochondrial electron transport. Plant J. Cell Mol. Biol. 2005, 41, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Modolo, L.V.; Augusto, O.; Almeida, I.M.; Magalhaes, J.R.; Salgado, I. Nitrite as the major source of nitric oxide production by arabidopsis thaliana in response to pseudomonas syringae. FEBS Lett. 2005, 579, 3814–3820. [Google Scholar] [CrossRef] [PubMed]
- Chamizo-Ampudia, A.; Sanz-Luque, E.; Llamas, A.; Ocana-Calahorro, F.; Mariscal, V.; Carreras, A.; Barroso, J.B.; Galvan, A.; Fernandez, E. A dual system formed by the arc and nr molybdoenzymes mediates nitrite-dependent no production in chlamydomonas. Plant Cell Environ. 2016, 39, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Luque, E.; Ocana-Calahorro, F.; de Montaigu, A.; Chamizo-Ampudia, A.; Llamas, A.; Galvan, A.; Fernandez, E. Thb1, a truncated hemoglobin, modulates nitric oxide levels and nitrate reductase activity. Plant J. Cell Mol. Biol. 2015, 81, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Rockel, P.; Strube, F.; Rockel, A.; Wildt, J.; Kaiser, W.M. Regulation of nitric oxide (NO) production by plant nitrate reductase in vivo and in vitro. J. Exp. Bot. 2002, 53, 103–110. [Google Scholar] [CrossRef]
- Chamizo-Ampudia, A.; Sanz-Luque, E.; Llamas, A.; Galvan, A.; Fernandez, E. Nitrate reductase regulates plant nitric oxide homeostasis. Trends Plant Sci. 2017, 22, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Savidov, N.A.; Tokarev, B.I.; Lips, S.H. Regulation of mo-cofactor, NADH- and NAD(P)H-specific nitrate reductase activities in the wild type and two nar-mutant lines of barley (Hordeum vulgare L.). J. Exp. Bot. 1997, 48, 847–855. [Google Scholar] [CrossRef]
- Wells, G.N.; Hageman, R.H. Specificity for nicotinamide adenine-dinucleotide by nitrate reductase from leaves. Plant Physiol. 1974, 54, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.V.; Harper, J.E. The conversion of nitrite to nitrogen oxide(s) by the constitutive NAD(P)H-nitrate reductase enzyme from soybean. Plant Physiol. 1988, 88, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sukumaran, S.; Marton, L. Differential expression of the arabidopsis NIA1 and NIA2 genes. Cytokinin-induced nitrate reductase activity is correlated with increased NIA1 transcription and mrna levels. Plant Physiol. 1998, 116, 1091–1096. [Google Scholar] [CrossRef]
- Cheng, C.L.; Acedo, G.N.; Dewdney, J.; Goodman, H.M.; Conkling, M.A. Differential expression of the two arabidopsis nitrate reductase genes. Plant Physiol. 1991, 96, 275–279. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, C.L. A chlorate-resistant mutant defective in the regulation of nitrate reductase gene expression in arabidopsis defines a new hy locus. Plant Cell 1997, 9, 21–35. [Google Scholar] [CrossRef]
- Wilkinson, J.Q.; Crawford, N.M. Identification of the arabidopsis chl3 gene as the nitrate reductase structural gene NIA2. Plant Cell 1991, 3, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Braaksma, F.J.; Feenstra, W.J. Isolation and characterization of nitrate reductase-deficient mutants of arabidopsis thaliana. Theor. Appl. Genet. 1982, 64, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.; Desikan, R.; Hancock, J.T.; Weir, I.S.; Neill, S.J. Aba-induced no generation and stomatal closure in arabidopsis are dependent on H2O2 synthesis. Plant J. Cell Mol. Biol. 2006, 45, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.S.; Zhao, S.L.; Dong, H.; Zhang, H.; Sun, L.R.; Miao, C. NIA1 and NIA2 are involved in exogenous salicylic acid-induced nitric oxide generation and stomata! Closure in arabidopsis. J. Integr. Plant Biol. 2010, 52, 298–307. [Google Scholar] [CrossRef]
- Kubo, Y.; Ogura, N.; Nakagawa, H. Limited proteolysis of the nitrate reductase from spinach leaves. J. Biol. Chem. 1988, 263, 19684–19689. [Google Scholar] [PubMed]
- Campbell, W.H. Nitrate reductase structure, function and regulation: Bridging the gap between biochemistry and physiology. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 277–303. [Google Scholar] [CrossRef] [PubMed]
- Solomonson, L.P.; Barber, M.J. Assimilatory nitrate reductase: Functional properties and regulation. Annu. Rev. Plant Phys. 1990, 41, 225–253. [Google Scholar] [CrossRef]
- Barber, M.J.; Notton, B.A. Spinach nitrate reductase: Effects of ionic strength and ph on the full and partial enzyme activities. Plant Physiol. 1990, 93, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Lambeck, I.C. Post-Translationale Regulation der Nitratreduktase Durch Phosphorylierung und 14-3-3-Protein-Bindung. Ph.D. Thesis, Universität zu Köln, Cologne, Germany, 2009. [Google Scholar]
- Lambeck, I.; Chi, J.C.; Krizowski, S.; Mueller, S.; Mehlmer, N.; Teige, M.; Fischer, K.; Schwarz, G. Kinetic analysis of 14-3-3-inhibited arabidopsis thaliana nitrate reductase. Biochemistry 2010, 49, 8177–8186. [Google Scholar] [CrossRef] [PubMed]
- Skipper, L.; Campbell, W.H.; Mertens, J.A.; Lowe, D.J. Pre-steady-state kinetic analysis of recombinant arabidopsis NADH:Nitrate reductase: Rate-limiting processes in catalysis. J. Biol. Chem. 2001, 276, 26995–27002. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.J.; James, D.M.; Eaglesham, A.R. The non-enzymic reduction of nitrite by benzyl viologen (free-radical) in the presence and absence of ammonium sulphate. Mol. Cell Biochem. 1975, 6, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.J.; Nason, A. Pyridine nucleotide-nitrate reductase from extracts of higher plants. Plant Physiol. 1953, 28, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Song, J.T.; Seo, H.S. Arabidopsis nitrate reductase activity is stimulated by the E3 SUMO ligase ATSIZ1. Nat. Commun. 2011, 2, 400. [Google Scholar] [CrossRef] [PubMed]
- Streit, L.; Nelson, R.S.; Harper, J.E. Nitrate reductases from wild-type and nr(1)-mutant soybean (Glycine max [L.] Merr.) Leaves: I. Purification, kinetics, and physical properties. Plant Physiol. 1985, 78, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.S.; Streit, L.; Harper, J.E. Nitrate reductases from wild-type and nr(1)-mutant soybean (Glycine max [L.] Merr.) Leaves: II. Partial activity, inhibitor, and complementation analyses. Plant Physiol. 1986, 80, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Jolly, S.O.; Campbell, W.; Tolbert, N.E. Nadph- and NADH-nitrate reductases from soybean leaves. Arch. Biochem. Biophys. 1976, 174, 431–439. [Google Scholar] [CrossRef]
- Wilkinson, J.Q.; Crawford, N.M. Identification and characterization of a chlorate-resistant mutant of arabidopsis thaliana with mutations in both nitrate reductase structural genes NIA1 and NIA2. Mol. Gen. Genet. 1993, 239, 289–297. [Google Scholar] [PubMed]
- Britto, D.T.; Kronzucker, H.J. Constancy of nitrogen turnover kinetics in the plant cell: Insights into the integration of subcellular n fluxes. Planta 2001, 213, 175–181. [Google Scholar] [CrossRef]
- Britto, D.T.; Kronzucker, H.J. The case for cytosolic no3- heterostasis: A critique of a recently proposed model. Plant Cell Environ. 2003, 26, 183–188. [Google Scholar] [CrossRef]
- Davenport, S.; Le Lay, P.; Sanchez-Tamburrrino, J.P. Nitrate metabolism in tobacco leaves overexpressing arabidopsis nitrite reductase. Plant Physiol. Biochem. 2015, 97, 96–107. [Google Scholar] [CrossRef]
- Miller, A.J.; Smith, S.J. Nitrate transport and compartmentation in cereal root cells. J. Exp. Bot. 1996, 47, 843–854. [Google Scholar] [CrossRef]
- Sugiura, M.; Georgescu, M.N.; Takahashi, M. A nitrite transporter associated with nitrite uptake by higher plant chloroplasts. Plant Cell Physiol. 2007, 48, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.M.; Desikan, R.; Bright, J.; Confraria, A.; Harrison, J.; Hancock, J.T.; Barros, R.S.; Neill, S.J.; Wilson, I.D. Differential requirement for no during aba-induced stomatal closure in turgid and wilted leaves. Plant Cell Environ. 2009, 32, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.D.; Ribeiro, D.M.; Bright, J.; Confraria, A.; Harrison, J.; Barros, R.S.; Desikan, R.; Neill, S.J.; Hancock, J.T. Role of nitric oxide in regulating stomatal apertures. Plant Signal. Behav. 2009, 4, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Barbier, G.G.; Campbell, W.H. Viscosity effects on eukaryotic nitrate reductase activity. J. Biol. Chem. 2005, 280, 26049–26054. [Google Scholar] [CrossRef] [PubMed]
- Krainer, F.W.; Capone, S.; Jäger, M.; Vogl, T.; Gerstmann, M.; Glieder, A.; Herwig, C.; Spadiut, O. Optimizing cofactor availability for the production of recombinant heme peroxidase in pichia pastoris. Microb. Cell Factor. 2015, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G.; Boxer, D.H.; Mendel, R.R. Molybdenum cofactor biosynthesis. The plant protein cnx1 binds molybdopterin with high affinity. J. Biol. Chem. 1997, 272, 26811–26814. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage t4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Lawrence, A.M.; Besir, H.U. Staining of proteins in gels with coomassie g-250 without organic solvent and acetic acid. J. Vis. Exp. 2009, 14, 1350. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]
- MacArthur, P.H.; Shiva, S.; Gladwin, M.T. Measurement of circulating nitrite and s-nitrosothiols by reductive chemiluminescence. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 851, 93–105. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | KMnitrite [µM] | kcatnitrite [s−1] |
|---|---|---|
| NIA1-Mo-heme | 35.5 ± 2.7 | 19.8 ± 4.8 |
| NIA1-Mo | 35.3 ± 3.2 | 137.5 ± 2.5 |
| NIA2-Mo-heme | 13.7 ± 3.3 | 1.8 ± 0.3 |
| NIA2-Mo-heme-H600A | 10.3 ± 1.2 | 1.2 ± 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohn, M.A.; Thaqi, B.; Fischer-Schrader, K. Isoform-Specific NO Synthesis by Arabidopsis thaliana Nitrate Reductase. Plants 2019, 8, 67. https://doi.org/10.3390/plants8030067
Mohn MA, Thaqi B, Fischer-Schrader K. Isoform-Specific NO Synthesis by Arabidopsis thaliana Nitrate Reductase. Plants. 2019; 8(3):67. https://doi.org/10.3390/plants8030067
Chicago/Turabian StyleMohn, Marie Agatha, Besarta Thaqi, and Katrin Fischer-Schrader. 2019. "Isoform-Specific NO Synthesis by Arabidopsis thaliana Nitrate Reductase" Plants 8, no. 3: 67. https://doi.org/10.3390/plants8030067
APA StyleMohn, M. A., Thaqi, B., & Fischer-Schrader, K. (2019). Isoform-Specific NO Synthesis by Arabidopsis thaliana Nitrate Reductase. Plants, 8(3), 67. https://doi.org/10.3390/plants8030067