Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species



Abstract
1. Introduction
2. Results
2.1. Variation in Organic Acid and Soluble Sugar Contents in the Mature Fruits of Two Wild Apple Species
2.2. Quality and Coverage of the Apple Fruit Proteome
2.3. Verification of the Differentially Expressed Proteins by PRM
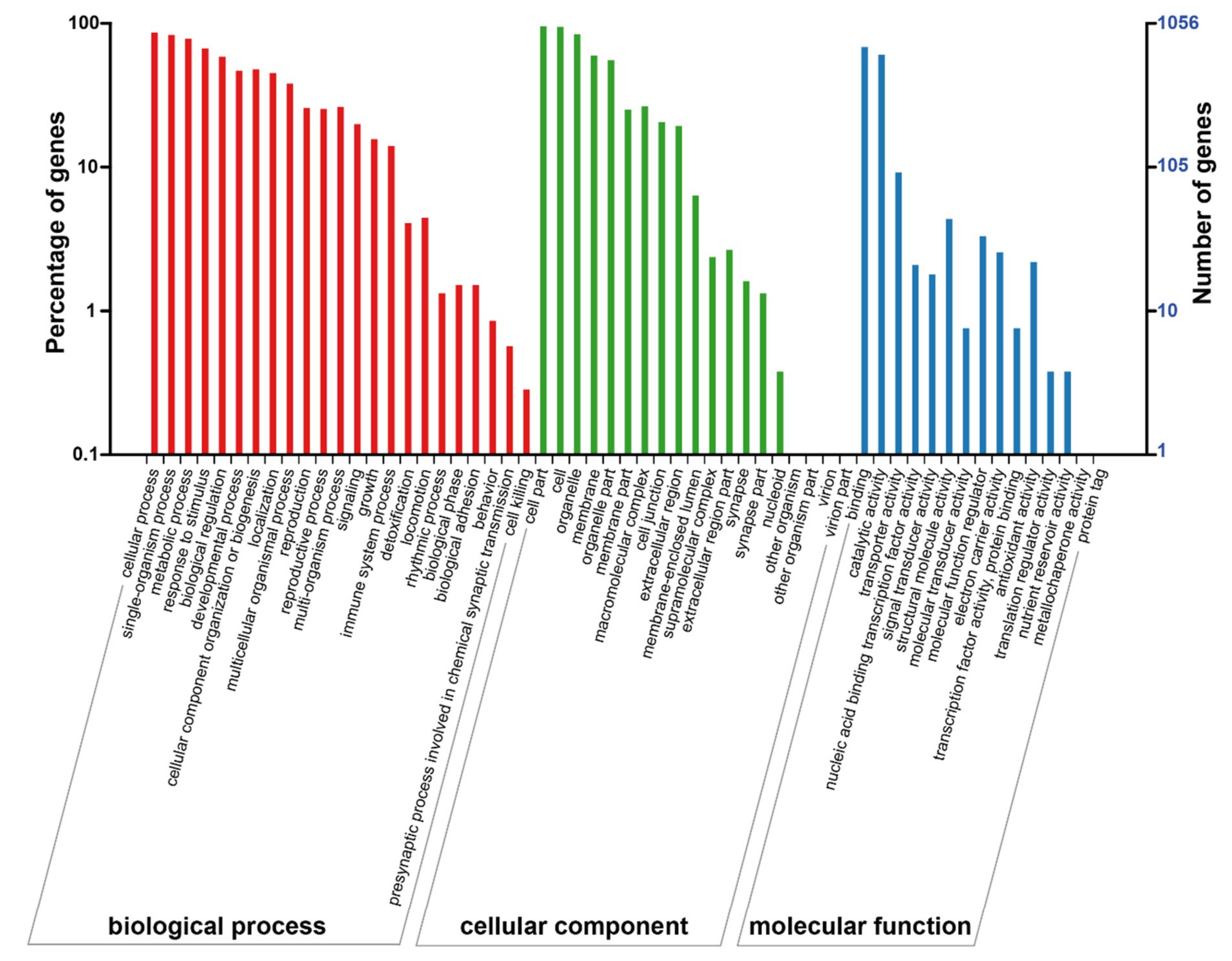
2.4. Analysis of Differentially-Expressed Proteins Based on the gene ontology (GO) and KEGG Databases
2.5. Specificity of the Protein Accumulation Related to Soluble Sugar and Organic Acid Metabolism
2.6. Identification of Proteins Involved in H+ transport
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Soluble Sugar and Organic Acid Contents
4.3. Protein Extraction, Enzymatic Hydrolysis, and Peptide Quantification
4.4. Data Analysis
4.5. Bioinformatic Analysis
4.6. PRM Verification
Supplementary Materials
Data Archiving Statement
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borsani, J.; Budde, C.O.; Porrini, L.; Lauxmann, M.A.; Lombardo, V.A.; Murray, R.; Andreo, C.S.; Drincovich, M.F.; Lara, M.V. Carbon metabolism of peach fruit after harvest: Changes in enzymes involved in organic acid and sugar level modifications. J. Exp. Bot. 2009, 60, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, J.; Zheng, H.; Fang, T.; Ogutu, C.; Li, S.; Han, Y.; Wu, B. Comparative assessment of sugar and malic acid composition in cultivated and wild apples. Food Chem. 2015, 172, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Gottwald, J.R.; Krysan, P.J.; Young, J.C.; Evert, R.F.; Sussman, M.R. Genetic evidence for the in planta role of phloem-specific plasma membrane sucrose transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13979–13984. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Q.; Fang, T.; Peng, Q.; Liao, L.; Zhao, L.; Owiti, A.; Han, Y. Developing gene-tagged molecular markers for evaluation of genetic association of apple SWEET genes with fruit sugar accumulation. Hortic. Res. 2018, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Diakou, P.; Svanella, L.; Raymond, P.; Gaudillère, J.P.; Moing, A. Phosphoenolpyruvate carboxylase during grape berry development: Protein level, enzyme activity and regulation. Aust. J. Plant. Physiol. 2000, 27, 221–229. [Google Scholar] [CrossRef]
- Mahmood, T.; Anwar, F.; Abbas, M.; Boyce, M.C.; Saari, N. Compositional variation in sugars and organic acids at different maturity stages in selected small fruits from Pakistan. Int. J. Mol. Sci. 2012, 13, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Liu, X.H.; Chen, L.S. Developmental changes in pulp organic acid concentration and activities of acid-metabolising enzymes during the fruit development of two loquat (Eriobotrya japonica Lindl.) cultivars differing in fruit acidity. Food Chem. 2009, 114, 657–664. [Google Scholar] [CrossRef]
- Wu, J.; Gao, H.; Zhao, L.; Liao, X.; Chen, F.; Wang, Z.; Hu, X. Chemical compositional characterization of some apple cultivars. Food Chem. 2007, 103, 88–93. [Google Scholar] [CrossRef]
- Lu, X.P.; Liu, Y.Z.; Zhou, G.F.; Wei, Q.J.; Hu, H.J.; Peng, S.A. Identification of organic acid-related genes and their expression profiles in two pear (Pyrus pyrofolia) cultivars with difference in predominant acid type at fruit ripening stage. Sci. Hortic. 2011, 129, 680–687. [Google Scholar] [CrossRef]
- Scherer, R.; Rybka, A.C.P.; Ballus, C.A.; Meinhart, A.D.; Filho, J.T.; Godoy, H.T. Validation of a HPLC method for simultaneous determination of main organic acids in fruits and juices. Food Chem. 2012, 135, 150–154. [Google Scholar] [CrossRef]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, Techniques and Possibilities to Overcome Biological Sample Complexity. Hum. Genom. Proteom. 2009, 2009, 239204. [Google Scholar] [CrossRef] [PubMed]
- Gallego, M.; Mora, L.; Aristoy, M.C.; Toldrá, F. Optimisation of a simple and reliable label-free methodology for the relative quantitation of raw pork meat proteins. Food Chem. 2015, 182, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Ulaszek, R.; Deciu, C.; Schieltz, D.M.; Yates, J.R. Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal. Chem. 2002, 74, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Chelius, D.; Bondarenko, P.V. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res. 2002, 1, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Quadroni, M.; Ducret, A.; Stöcklin, R. Quantify this! Report on a round table discussion on quantitative mass spectrometry in proteomics. Proteomics 2004, 4, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Burlingame, A.L.; Bradshaw, R.A. Western blots versus selected reaction monitoring assays: Time to turn the tables? Mol. Cell. Proteom. 2013, 12, 2381–2382. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Zhang, X.J.; Zhao, Z.Y. Effects of fruit bagging on anthocyanins, sugars, organic acids, and color properties of ‘Granny Smith’ and ‘Golden Delicious’ during fruit maturation. Eur. Food Res. Technol. 2013, 236, 329–339. [Google Scholar] [CrossRef]
- Hecke, K.; Herbinger, K.; Veberic, R.; Trobec, M.; Toplak, H.; Stampar, F.; Keppel, H.; Grill, D. Sugar-, acid- and phenol contents in apple cultivars from organic and integrated fruit cultivation. Eur. J. Clin. Nutr. 2006, 60, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Petkovsek, M.M.; Stampar, F.; Veberic, R. Parameters of inner quality of the apple scab resistant and susceptible apple cultivars (Malus domestica Borkh.). Sci. Hortic. 2007, 114, 37–44. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Cheng, L. Developmental changes of carbohydrates, organic acids, amino acids, and phenolic compounds in ‘Honeycrisp’ apple flesh. Food Chem. 2010, 123, 1013–1018. [Google Scholar] [CrossRef]
- Ma, B.Q.; Yuan, Y.Y.; Gao, M.; Li, C.Y.; Ogutu, C.; Li, M.J.; Ma, F.W. Determination of Predominant Organic Acid Components in Malus Species: Correlation with Apple Domestication. Metabolites 2018, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, D.; Feng, F.; Zhang, S.; Ma, F.; Cheng, L. Proteomic analysis reveals dynamic regulation of fruit development and sugar and acid accumulation in apple. J. Exp. Bot. 2016, 67, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, C.; Deluc, L.G.; Cramer, G.R.; Ford, C.M.; Soole, K.L. Regulation of malate metabolism in grape berry and other developing fruits. Phytochemistry 2009, 70, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Etienne, A.; Génard, M.; Lobit, P.; Mbeguié-A-Mbéguié, D.; Bugaud, C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 2013, 64, 145–169. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Boo, K.H.; Kim, H.Y.; Eigenheer, R.A.; Phinney, B.S.; Shulaev, V.; Negre-Zakharov, F.; Sadka, A.; Blumwald, E. Label-free shotgun proteomics and metabolite analysis reveal a significant metabolic shift during citrus fruit development. J. Exp. Bot. 2011, 62, 5367–5384. [Google Scholar] [CrossRef] [PubMed]
- Sadka, A.; Dahan, E.; Or, E.; Roose, M.L.; Marsh, K.B.; Cohen, L. Comparative analysis of mitochondrial citrate synthase gene structure, transcript level and enzymatic activity in acidless and acid-containing Citrus varieties. Funct. Plant. Biol. 2001, 28, 383–390. [Google Scholar] [CrossRef]
- Hafke, J.B.; Hafke, Y.; Smith, J.A.; Lüttge, U.; Thiel, G. Vacuolar malate uptake is mediated by an anion-selective inward rectifier. Plant. J. 2003, 35, 116–128. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Fang, X.; Zhu, T.; Han, S.; Zhu, H.; Li, S. Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin. Metabolites 2019, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvás, P.; Prášil, I.T.; Renaut, J. Plant proteome changes under abiotic stress--contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.; Zhang, Y.; Dai, S. Mechanisms of plant salt response: Insights from proteomics. J. Proteome Res. 2012, 11, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Smaczniak, C.; de Vries, S.; Angenent, G.C.; Karlova, R. Proteomics insights into plant signaling and development. Proteomics 2011, 11, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.M.; Corpas, F.J.; del Río, L.A. Proteomics as an approach to the understanding of the molecular physiology of fruit development and ripening. J. Proteom. 2011, 74, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, J.; Levin, Y.; Savidor, A.; Breitel, D.; Chappell-Maor, L.; Heinig, U.; Töpfer, N.; Aharoni, A. Label-free deep shotgun proteomics reveals protein dynamics during tomato fruit tissues development. Plant. J. 2017, 90, 396–417. [Google Scholar] [CrossRef] [PubMed]
- Baerenfaller, K.; Grossmann, J.; Grobei, M.A.; Hull, R.; Hirsch-Hoffmann, M.; Yalovsky, S.; Zimmermann, P.; Grossniklaus, U.; Gruissem, W.; Baginsky, S. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 2008, 320, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Mintz-Oron, S.; Mandel, T.; Rogachev, I.; Feldberg, L.; Lotan, O.; Yativ, M.; Wang, Z.; Jetter, R.; Venger, I.; Adato, A.; et al. Gene expression and metabolism in tomato fruit surface tissues. Plant. Physiol. 2008, 147, 823–851. [Google Scholar] [CrossRef] [PubMed]
- Famiani, F.; Battistelli, A.; Moscatello, S.; Cruz-Castillo, J.G.; Walker, R.P. The organic acids that are accumulated in the flesh of fruits: Occurrence, metabolism and factors affecting their contents-a review. Rev. Chapingo Ser. Hortic. 2015, 21, 97–128. [Google Scholar] [CrossRef]
- Hockema, B.R.; Etxeberria, E. Metabolic contributors to drought-enhanced accumulation of sugars and acids in oranges. J. Am. Soc. Hortic. Sci. 2001, 126, 599–605. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, L.; Cui, W.; Zhang, C.; Li, D.; Ma, B.; Cheng, L.; Ruan, Y.L.; Ma, F.; Li, M. Increased activity of MdFRK2, a high-affinity fructokinase, leads to upregulation of sorbitol metabolism and downregulation of sucrose metabolism in apple leaves. Hortic. Res. 2018, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Givan, C.V. Evolving concepts in plant glycolysis: Two centuries of progress. Biol. Rev. 1999, 74, 277–309. [Google Scholar] [CrossRef]
- Yao, Y.X.; Li, M.; Zhai, H.; You, C.X.; Hao, Y.J. Isolation and characterization of an apple cytosolic malate dehydrogenase gene reveal its function in malate synthesis. J. Plant. Physiol. 2011, 168, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Pua, E.C.; Chandramouli, S.; Han, P.; Liu, P. Malate synthase gene expression during fruit ripening of Cavendish banana (Musa acuminata cv. Williams). J. Exp. Bot. 2003, 54, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Terrier, N.; Glissant, D.; Grimplet, J.; Barrieu, F.; Abbal, P.; Couture, C.; Ageorges, A.; Atanassova, R.; Léon, C.; Renaudin, J.P.; et al. Isogene specific oligo arrays reveal multifaceted changes in gene expression during grape berry (Vitis vinifera L.) development. Planta 2005, 222, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Martinoia, E.; Maeshima, M.; Neuhaus, H.E. Vacuolar transporters and their essential role in plant metabolism. J. Exp. Bot. 2007, 58, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Liao, L.; Fang, T.; Peng, Q.; Ogutu, C.; Zhou, H.; Ma, F.; Han, Y. A Ma10 gene encoding P-type ATPase is involved in fruit organic acid accumulation in apple. Plant. Biotechnol. J. 2019, 17, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Etienne, C.; Moing, A.; Dirlewanger, E.; Raymond, P.; Monet, R.; Rothan, C. Isolation and characterization of six peach cDNAs encoding key proteins in organic acid metabolism and solute accumulation: Involvement in regulating peach fruit acidity. Physiol. Plant. 2002, 114, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.T.; Xie, C.Y.; Jiang, H.X.; Chen, L.S. Expression of six malate-related genes in pulp during the fruit development of two loquat (Eriobotrya japonica) cultivars differing in fruit acidity. Afr. J. Biotechnol. 2011, 10, 2414–2422. [Google Scholar]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of largescale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Results | Unique Peptides | Quantified Proteins | Msa vs. Mni | Significantly Different Proteins | |
|---|---|---|---|---|---|
| Up-Regulated | Down-Regulated | ||||
| Total | 13036 | 2901 | 416 | 663 | 1079 |
| Peptide Sequence | Protein Name | Average Content | PRM Fold Change | Label-Free Fold Change | Consistency between PRM and Label-Free | |
|---|---|---|---|---|---|---|
| Msa | Mni | |||||
| EYYTNALAAAK | MDP0000271088 | 0.0127 | 0.1152 | 0.1106 | 0.7260 | Yes |
| VSLGNFPDLAGAVNK | ||||||
| FLVSDSFPGNDR | MDP0000152497 | 0.6065 | 0.7086 | 0.8559 | 0.3358 | Yes |
| LVPIINPTTR | ||||||
| INDQAGYSSFR | MDP0000326249 | 0.4397 | 0.0483 | 9.1035 | 2.9726 | Yes |
| LANILHANELAR | ||||||
| ANEAALDLVR | MDP0000777702 | 0.3039 | / | _ | _ | Yes |
| YNEGALPGFDPATK | ||||||
| AFVDSGAQSTIISK | MDP0000308185 | / | 0.2294 | _ | _ | Yes |
| GIAHGVGQSEILGR | ||||||
| LVELGFGR | ||||||
| Item | Value |
|---|---|
| Enzyme | Trypsin |
| Max missed cleavages | 2 |
| Max missed cleavages | 2 |
| Main search | 6 ppm |
| First search | 20 ppm |
| MS/MS tolerance | 20 ppm |
| Fixed modifications | Carbamidomethyl (C) |
| Variable modifications | Oxidation (M), acetyl (protein N-term) |
| Peptide FDR | ≤0.01 |
| Protein FDR | ≤0.01 |
| Time window (match between runs) | 2 min |
| Protein quantification | Razor and unique peptides were used for protein quantification. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, B.; Ding, Y.; Li, C.; Li, M.; Ma, F.; Yuan, Y. Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species. Plants 2019, 8, 488. https://doi.org/10.3390/plants8110488
Ma B, Ding Y, Li C, Li M, Ma F, Yuan Y. Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species. Plants. 2019; 8(11):488. https://doi.org/10.3390/plants8110488
Chicago/Turabian StyleMa, Baiquan, Yuduan Ding, Cuiying Li, Mingjun Li, Fengwang Ma, and Yangyang Yuan. 2019. "Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species" Plants 8, no. 11: 488. https://doi.org/10.3390/plants8110488
APA StyleMa, B., Ding, Y., Li, C., Li, M., Ma, F., & Yuan, Y. (2019). Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species. Plants, 8(11), 488. https://doi.org/10.3390/plants8110488