
Combined Analysis of Transcriptome and Metabolome Reveals the Heat Stress Resistance of Dongxiang Wild Rice at Seedling Stage

 ,
,
Abstract
1. Introduction
2. Results
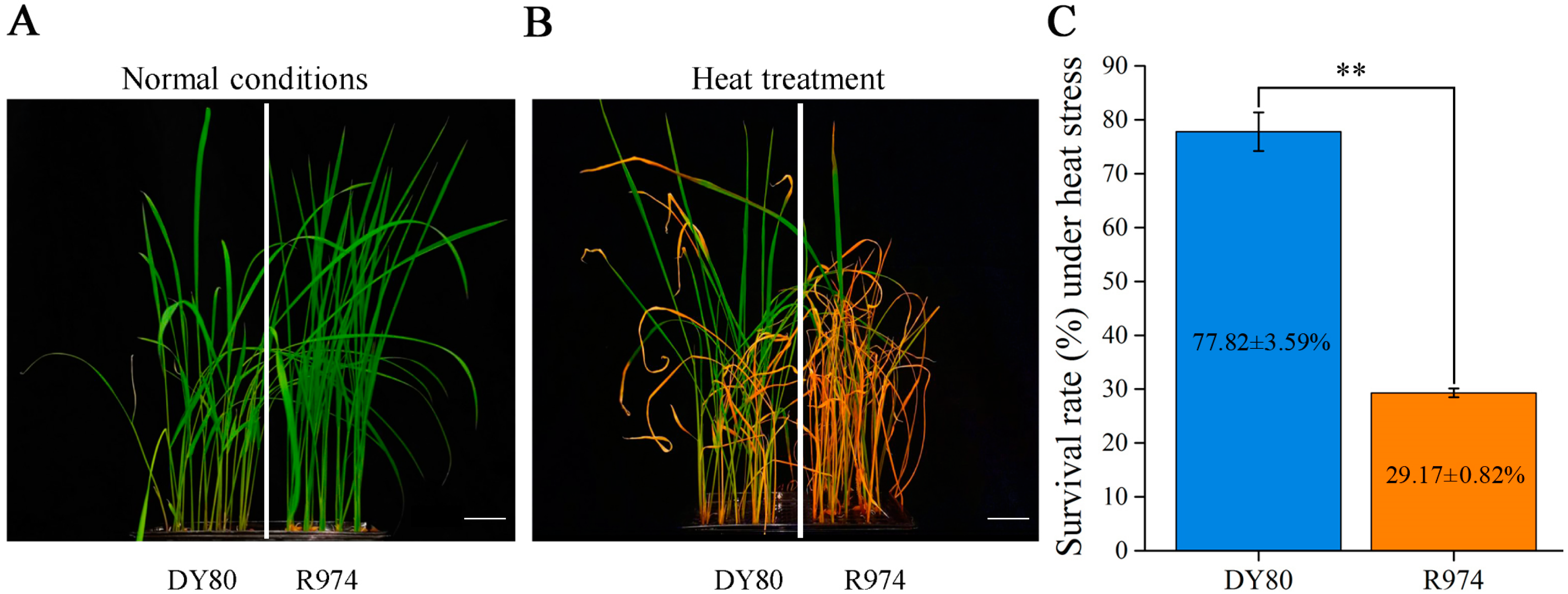
2.1. Performance of DY80 and R974 Under Heat Stress
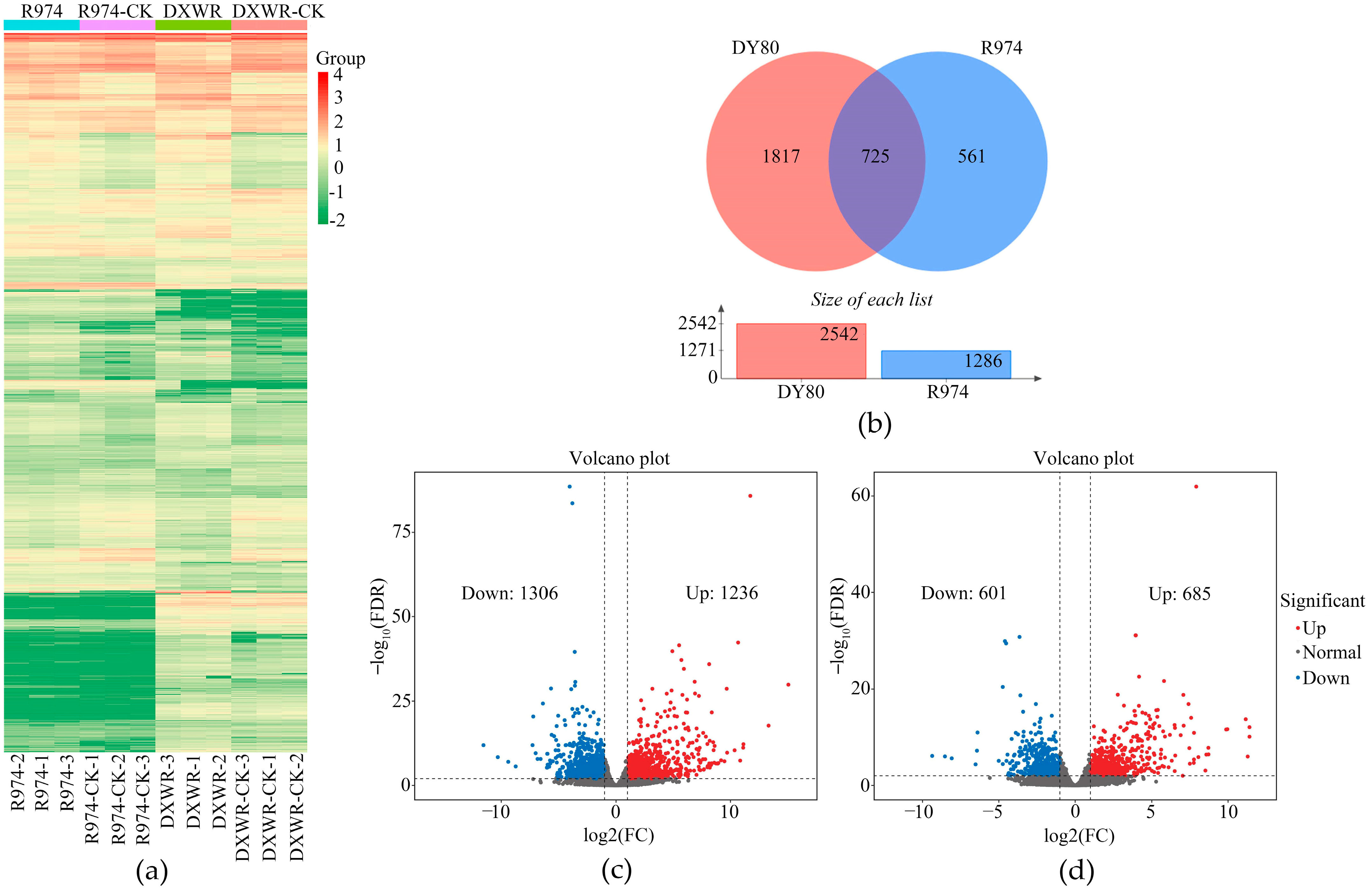
2.2. Transcriptome Sequencing
2.3. Gene Expression Patterns in DY80 and R974 After Heat Treatment
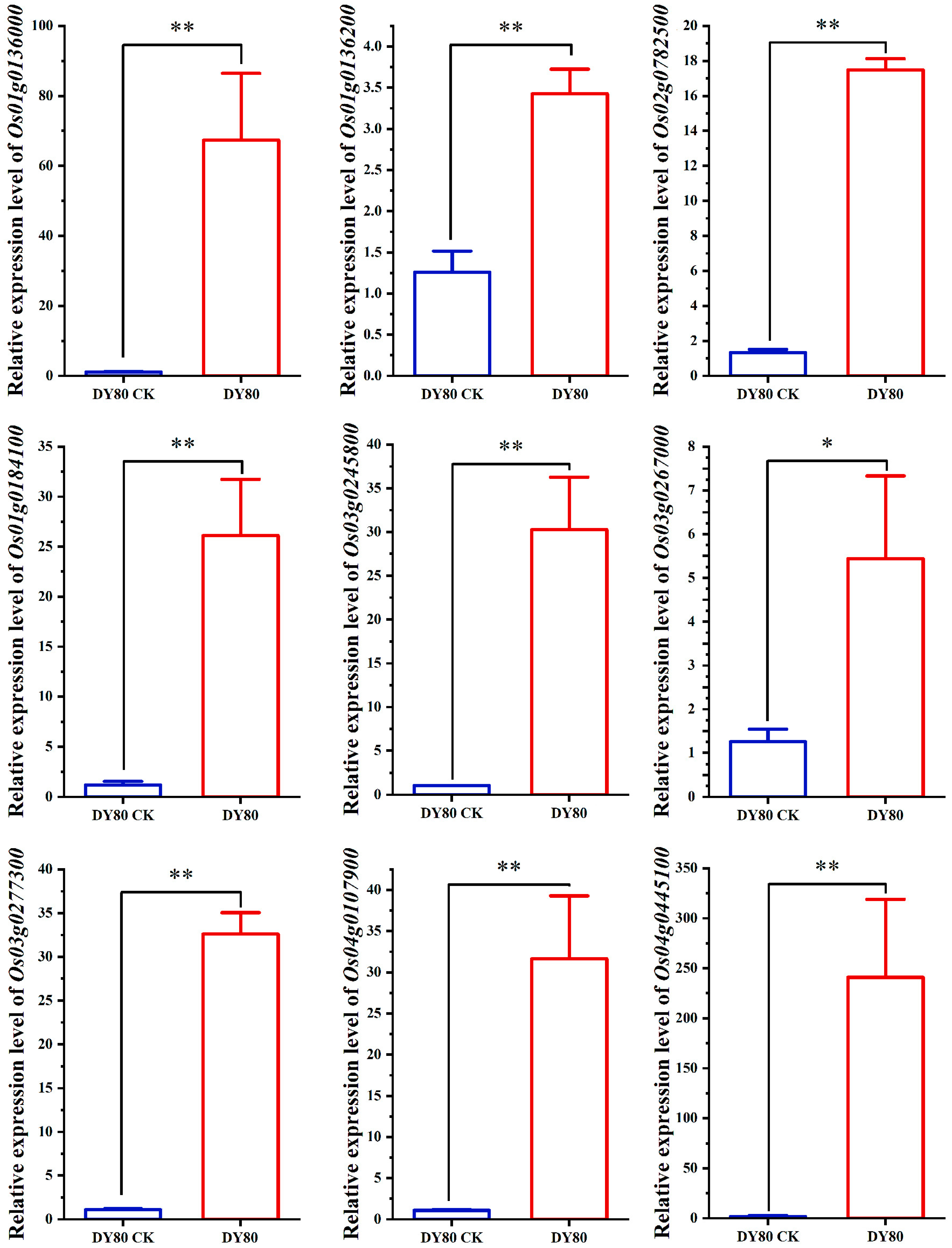
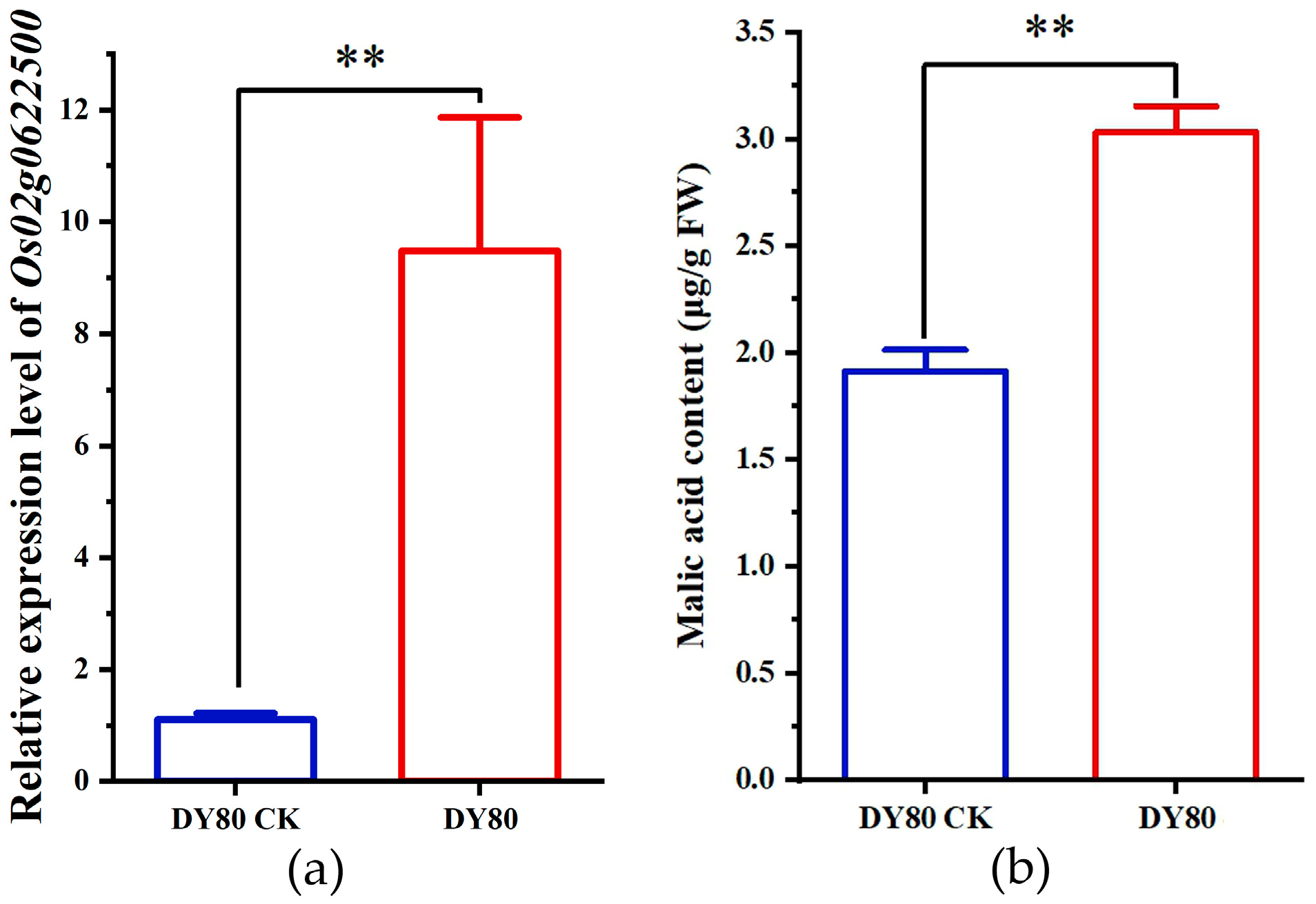
2.4. Verification of Transcriptomic Sequencing by qRT-PCR
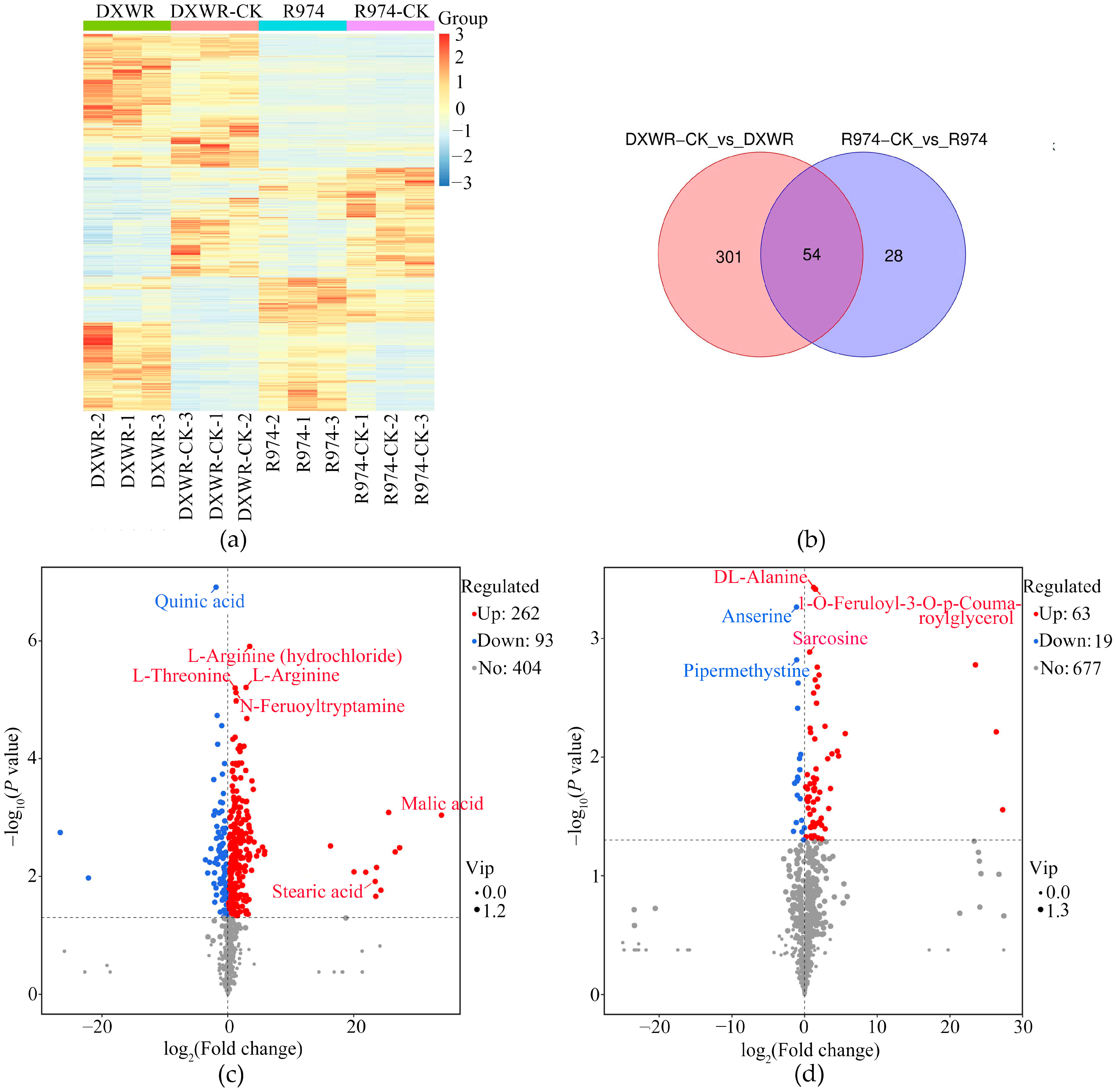
2.5. Analyses of Differential Metabolites After Heat Treatment in DY80 and R974
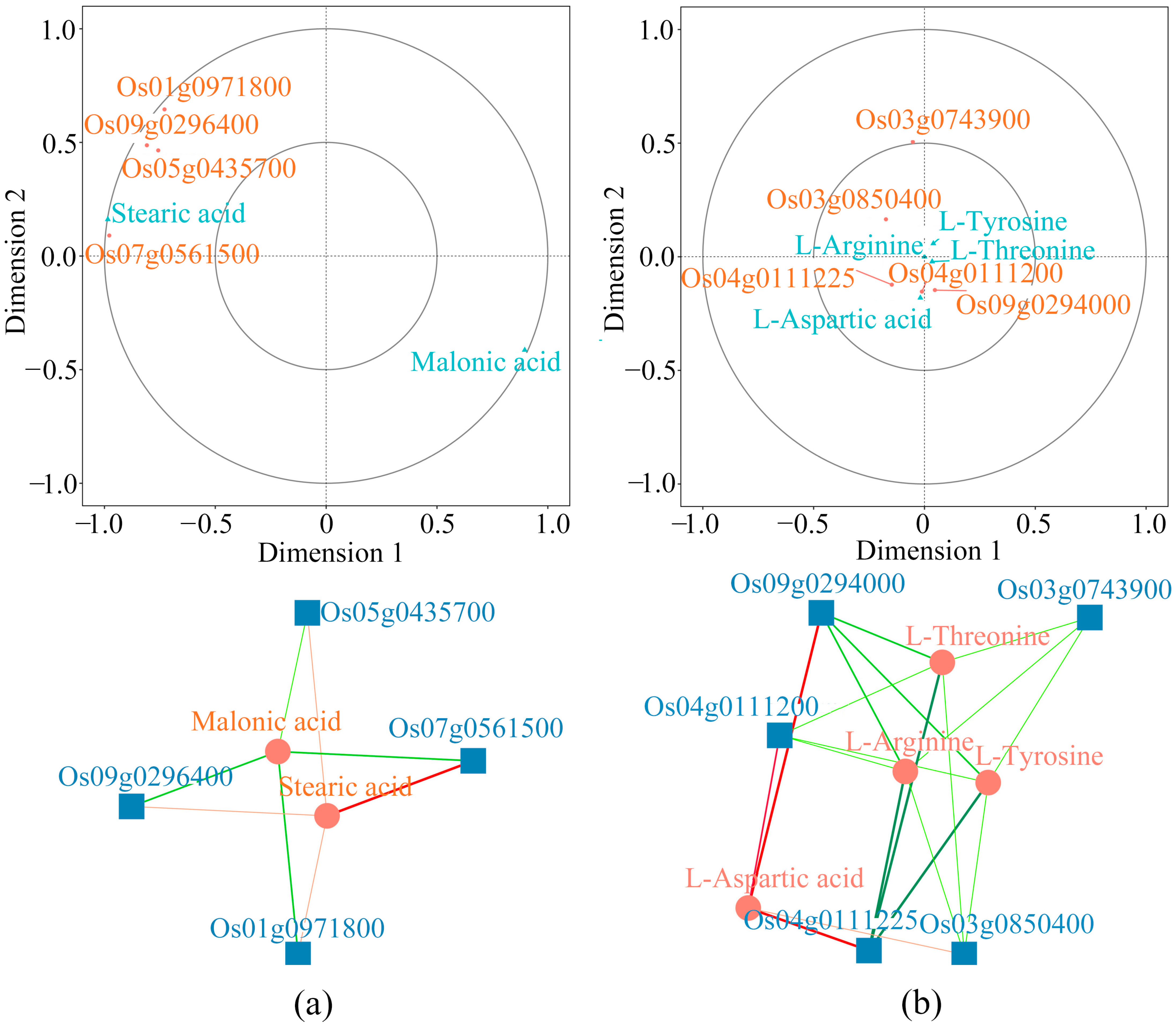
2.6. Combined Analyses of Differential Metabolites and DEGs
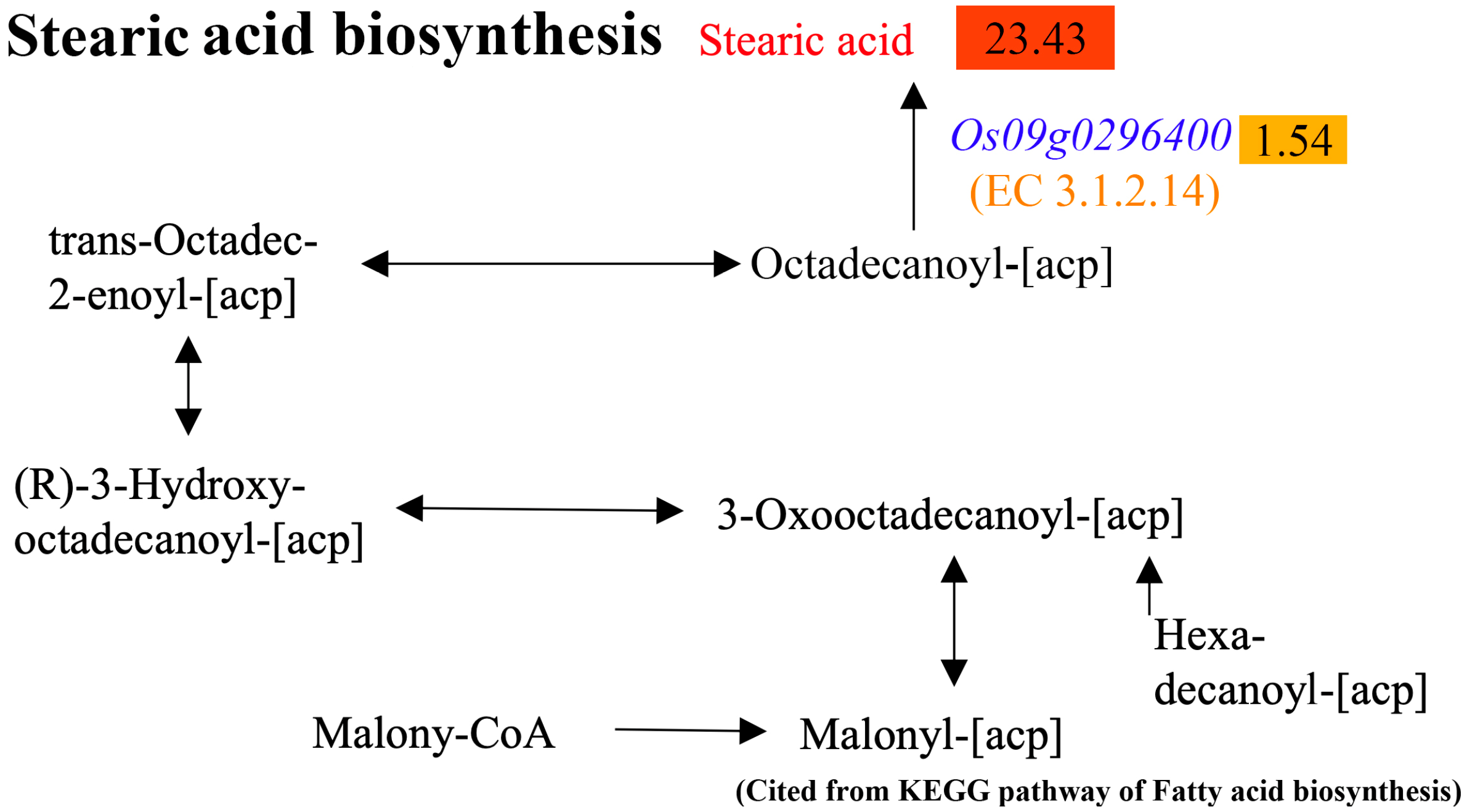
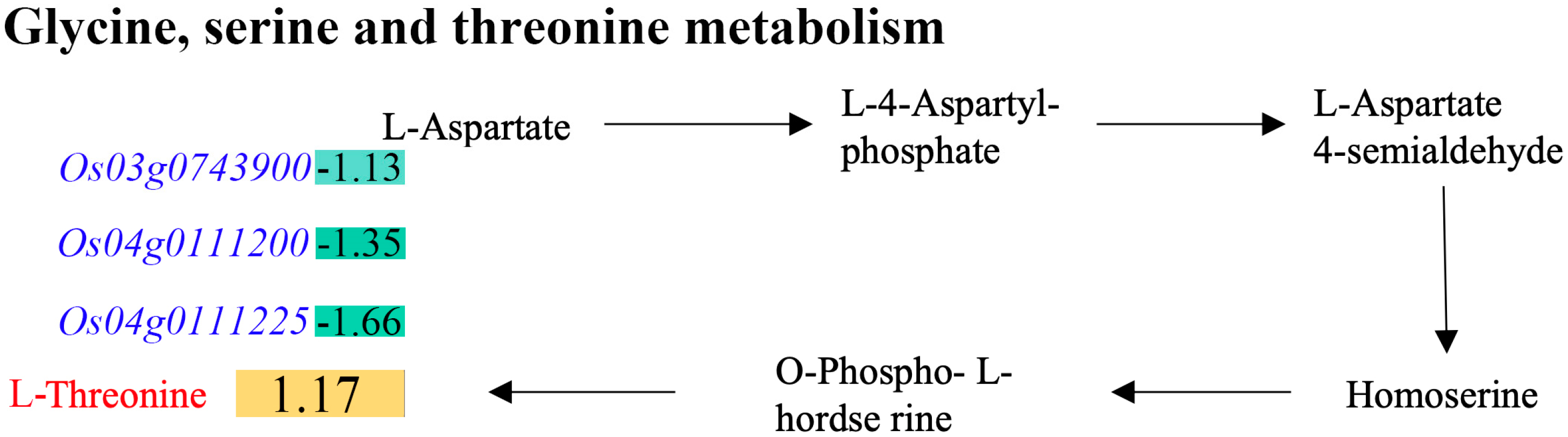
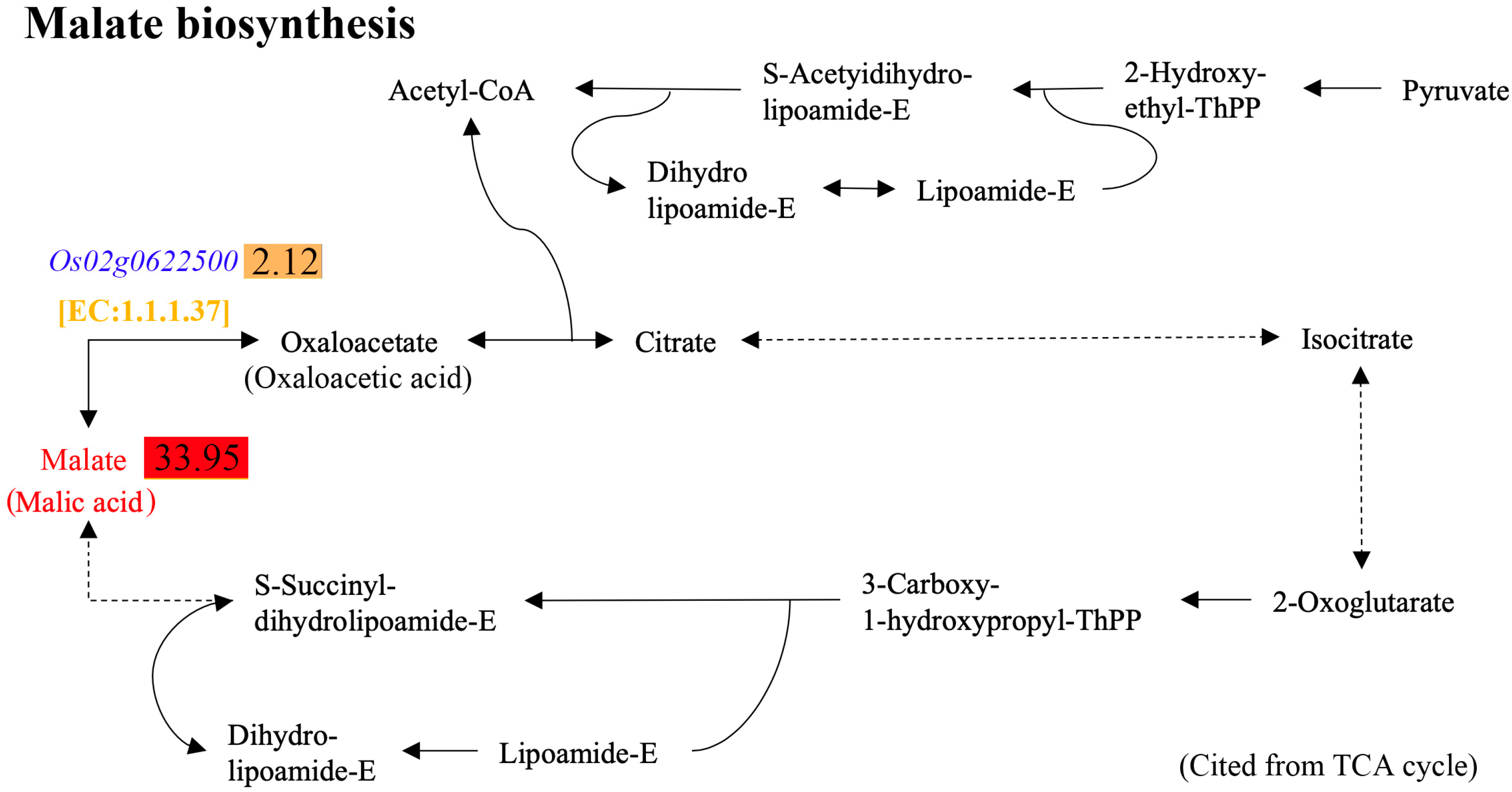
2.7. Verification of the Association Between Transcriptome and Metabolome Analyses
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Sample Preparation
4.3. Transcriptomic Sequencing
4.4. Metabolomic Analysis
4.5. Statistical Analyses
4.6. Real-Time Quantitative PCR
4.7. Verifying the Result of Combined Transcriptomic and Metabolomic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COG | Clusters of orthologous groups of proteins |
| DEGs | Differential expressed genes |
| EC | Enzyme classification |
| FC | Fold change |
| FDR | False discovery rate |
| GO | Gene ontology |
| GSEA | Gene set enrichment analysis |
| IPCC | Intergovernmental panel on climate change |
| KEGG | Kyoto encyclopedia of genes and genomes |
| KO | KEGG ortholog database |
| Nr | NCBI non-redundant protein sequences |
| Nt | NCBI non-redundant nucleotide sequences |
| PCA | Principal component analysis |
| VIP | Variable importance in projection |
| Pfam | Protein family |
| qRT-PCR | Real-time quantitative PCR |
| ROS | Reactive oxygen species |
| TCA | Tricarboxylic acid tricarboxylic acid |
| UPLC-MS/MS | Ultra-performance liquid chromatography–tandem mass spectrometry |
References
- Li, X.M.; Chao, D.Y.; Wu, Y.; Huang, X.; Chen, K.; Cui, L.G.; Su, L.; Ye, W.W.; Chen, H.; Chen, H.C.; et al. Natural alleles of a proteasome α2 subunit gene contribute to thermotolerance and adaptation of African rice. Nat. Genet. 2015, 47, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Huang, M.; Yao, Y.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Blumwald, E. Genetic engineering for modern agriculture: Challenges and perspectives. Annu. Rev. Plant Biol. 2010, 61, 443–462. [Google Scholar] [CrossRef]
- Gao, J.; Wallis, J.G.; Browse, J. Mutations in the prokaryotic pathway rescue the fatty acid biosynthesis1 mutant in the cold. Plant Physiol. 2015, 169, 442. [Google Scholar] [CrossRef] [PubMed]
- Rawat, N.; Singla-Pareek, S.L.; Pareek, A. Membrane dynamics during individual and combined abiotic stresses in plants and tools to study the same. Physiol. Plant. 2021, 171, 653–676. [Google Scholar] [CrossRef]
- Ding, Y.; Shi, Y.; Yang, S. Molecular regulation of plant responses to environmental temperatures. Mol. Plant 2020, 13, 544–564. [Google Scholar] [CrossRef]
- Dwivedi, S.K.; Basu, S.; Kumar, S.; Kumari, S.; Kumar, A.; Jha, S.; Mishra, J.S.; Bhatt, B.P.; Kumar, G. Enhanced antioxidant enzyme activities in developing anther contributes to heat stress alleviation and sustains grain yield in wheat. Funct. Plant Biol. 2019, 46, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Matsuba, S.; Funatsuki, H.; Kawaguchi, K.; Saruyama, H.; Tanida, M.; Sato, Y. Over-expression of a small heat shock protein, sHSP17. 7, confers both heat tolerance and UV-B resistance to rice plants. Mol. Breed. 2004, 13, 165–175. [Google Scholar] [CrossRef]
- El-Kweeamy, A.; Bi, Y.M.; Ranathunge, K.; Beatty, P.H.; Good, A.G.; Rothstein, S.J. The rice R2R3-MYB transcription factor OsMYB55 is involved in the tolerance to high temperature and modulates amino acid metabolism. PLoS ONE 2012, 7, e52030. [Google Scholar]
- Wang, H.; Bian, M.; Yang, Z.; Lin, C.; Shi, W. Preliminary functional analysis of the isoforms of OsHsfA2a (Oryza sativa L.) generated by alternative splicing. Plant Mol. Biol. Rep. 2013, 31, 38–46. [Google Scholar] [CrossRef]
- Lin, M.; Chai, K.; Ko, S.; Kuang, L.; Lur, H.S.; Charng, Y. A positive feedback loop between HEAT SHOCK PROTEIN101 and HEAT STRESS-ASSOCIATED 32-KD PROTEIN modulates long-term acquired thermotolerance illustrating diverse heat stress responses in rice varieties. Plant Physiol. 2014, 164, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, Z.; Liu, L.; Zhang, G.; Song, Z.; Guo, W.; Wang, X.; Qu, Y.; Zhu, Y.; Yang, D. An alternatively spliced heat shock transcription factor, OsHSFA2dI, functions in the heat stress-induced unfolded protein response in rice. Plant Biol. 2015, 17, 419–429. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, K.; Du, H.; Xu, Y.; Song, H.; Li, X.; Xiong, L. A stress-responsive NAC transcription factor SNAC3 confers heat and drought tolerance through modulation of reactive oxygen species in rice. J. Exp. Bot. 2015, 66, 6803–6817. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wei, C.; Liu, X.; Wang, M.; Yu, F.; Xie, Q.; Tu, J. The RING finger ubiquitin E3 ligase OsHTAS enhances heat tolerance by promoting H2O2-induced stomatal closure in rice. Plant Physiol. 2016, 170, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, L.; Ou, S.; Wang, R.; Wang, Y.; Chu, C.; Yao, S. Natural variations of SLG1 confer high-temperature tolerance in indica rice. Nat. Commun. 2020, 11, 5441. [Google Scholar] [CrossRef]
- Kan, Y.; Mu, X.R.; Zhang, H.; Gao, J.; Shan, J.X.; Ye, W.W.; Lin, H.X. TT2 controls rice thermotolerance through SCT1-dependent alteration of wax biosynthesis. Nat. Plants 2021, 8, 53–67. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, J.F.; Kan, Y.; Shan, J.X.; Ye, W.W.; Dong, N.Q.; Guo, T.; Xiang, Y.H.; Yang, Y.B.; Li, Y.C.; et al. A genetic module at one locus in rice protects chloroplasts to enhance thermotolerance. Science 2022, 376, 1293–1300. [Google Scholar] [CrossRef]
- Yang, C.; Luo, A.; Lu, H.; Davis, S.J.; Liu, J. Diurnal regulation of alternative splicing associated with thermotolerance in rice by two glycine-rich RNA-binding proteins. Sci. Bull. 2024, 69, 59–71. [Google Scholar] [CrossRef]
- Yamakawa, H.; Hakata, M. Atlas of rice grain filling-related metabolism under high temperature: Joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant Cell Physiol. 2010, 51, 795–809. [Google Scholar] [CrossRef]
- Lu, J.; Ye, R.; Qu, M.; Wang, Y.; Liang, T.; Lin, J.; Xie, R.; Ke, Y.; Gao, J.; Li, C.; et al. Combined transcriptome and proteome analysis revealed the molecular regulation mechanisms of zinc homeostasis and antioxidant machinery in tobacco in response to different zinc supplies. Plant Physiol. Biochem. 2023, 202, 107919. [Google Scholar] [CrossRef]
- Xiang, N.; Hu, J.; Yan, S.; Guo, X. Plant hormones and volatiles response to temperature stress in sweet corn (Zea mays L.) seedlings. J. Agric. Food Chem. 2021, 69, 6779–6790. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, X.; Zhu, S.; Zhang, D.; Han, S.; Li, Z.; Lu, J.; Chu, H.; Xiao, J.; Li, S. Integrated flow cytometric and proteomics analyses reveal the regulatory network underlying sugarcane protoplast responses to fusion. Plant Physiol. Biochem. 2023, 202, 107918. [Google Scholar] [CrossRef]
- Chen, C.; Xiang, J.; Yuan, J.; Shao, S.; Rehman, M.; Peng, D.; Liu, L. Comparative biochemical and transcriptomic analysis reveals the phosphate-starving tolerance of two ramie varieties. Plant Physiol. Biochem. 2023, 202, 107979. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.S.; Zheng, J.; Yan, C.; Li, X.X.; Liu, S.F.; Zhou, J.J.; Qin, X.J.; Nan, W.B.; Yang, Y.Q.; Zhang, H.M. Locating QTLs controlling overwintering trait in Chinese perennial Dongxiang wild rice. Mol. Genet. Genom. 2018, 293, 81–93. [Google Scholar] [CrossRef]
- Zhang, F.T.; Luo, X.D.; Zhou, Y.; Xie, J.K. Genome-wide identification of conserved microRNA and their response to drought stress in Dongxiang wild rice (Oryza rufipogon Griff.). Biotechnol. Lett. 2016, 38, 711–721. [Google Scholar] [CrossRef]
- Nie, Y.; Xia, H.; Ma, X.; Lou, Q.; Liu, Y.; Zhang, A.; Chen, L.; Yan, L.; Luo, L. Dissecting genetic basis of deep rooting in Dongxiang wild rice. Rice Sci. 2022, 29, 277–287. [Google Scholar]
- Qi, W.D.; Chen, H.P.; Yang, Z.Z.; Hu, B.L.; Luo, X.D.; Ai, B.; Luo, Y.; Huang, Y.; Xie, J.K.; Zhang, F.T. Systematic characterization of long non-coding RNAs and their responses to drought stress in Dongxiang wild rice. Rice Sci. 2020, 27, 21–31. [Google Scholar]
- Zhang, X.; Zhou, S.X.; Fu, Y.C.; Su, Z.; Wang, X.K.; Sun, C.Q. Identification of a drought tolerant introgression line derived from Dongxiang common wild rice (O. rufipogon Griff.). Plant Mol. Biol. 2006, 62, 247–259. [Google Scholar] [CrossRef]
- Zhao, F.; Luo, Y.; Zhang, M.; Zhou, Y.; Chen, H.; Hu, B.; Xie, J. Identification and characterization of drought stress responsive novel microRNAs in Dongxiang wild rice. Rice Sci. 2018, 25, 175–184. [Google Scholar]
- Li, F.; Guo, S.; Zhao, Y.; Chen, D.; Chong, K.; Xu, Y. Overexpression of a homopeptide repeat-containing bHLH protein gene (OrbHLH001) from Dongxiang Wild Rice confers freezing and salt tolerance in transgenic Arabidopsis. Plant Cell Rep. 2010, 29, 977–986. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, J.; Somg, Q.; Sun, C.; Liu, F. Combining QTL mapping and expression profile analysis to identify candidate genes of cold tolerance from Dongxiang common wild rice (Oryza rufipogon Griff.). J. Integr. Agr. 2016, 15, 1933–1943. [Google Scholar] [CrossRef]
- Erkina, T.Y.; Zou, Y.; Freeling, S.; Vorobyev, V.I.; Erkine, A.M. Functional interplay between chromatin remodeling complexes RSC, SWI/SNF and ISWI in regulation of yeast heat shock genes. Nucleic Acids Res. 2010, 38, 1441–1449. [Google Scholar] [CrossRef]
- Klopf, E.; Paskova, L.; Solé, C.; Mas, G.; Petryshyn, A.; Posas, F.; Wintersberger, U.; Ammerer, G.; Schüller, C. Cooperation between the INO80 complex and histone chaperones determines adaptation of stress gene transcription in the yeast Saccharomyces cerevisiae. Mol. Cell Biol. 2009, 29, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Venters, B.J.; Wachi, S.; Mavrich, T.; Andersen, B.; Jena, P.; Sinnamon, A.; Jain, P.; Rolleri, N.; Jiang, C.; Hemeryck-Walsh, C.; et al. A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol. Cell 2011, 41, 480–492. [Google Scholar] [CrossRef]
- Grams, T.; Lautner, S.; Felle, H.; Matyssek, R.; Fromm, J. Heat-induced electrical signals affect cytoplasmic and apoplastic pH as well as photosynthesis during propagation through the maize leaf. Plant Cell Environ. 2009, 32, 319–326. [Google Scholar] [CrossRef]
- He, Y.; Guan, H.; Li, B.; Zhang, S.; Xu, Y.; Yao, Y.; Yang, X.; Zha, Z.; Guo, Y.; Jiao, C.; et al. Transcriptome analysis reveals the dynamic and rapid transcriptional reprogramming involved in heat stress and identification of heat response genes in rice. Int. J. Mol. Sci. 2023, 24, 14802. [Google Scholar] [CrossRef]
- Tewari, A.K.; Tripathy, B.C. Temperature-stress-induced impairment of chlorophyll biosynthetic reactions in cucumber and wheat. Plant Physiol. 1998, 117, 851–858. [Google Scholar] [CrossRef]
- Obaid, A.Y.; Sabir, J.S.M.; Atef, A.; Liu, X.; Edris, S.; El-Domyati, F.M.; Mutwakil, M.Z.; Gadalla, N.O.; Hajrah, N.H.; Al-Kordy, M.A.; et al. Analysis of transcriptional response to heat stress in Rhazya stricta. BMC Plant Biol. 2016, 16, 252. [Google Scholar] [CrossRef]
- Sun, Y.; Geng, Q.; Du, Y.; Yang, X.; Zhai, H. Induction of cyclic electron flow around photosystem I during heat stress in grape leaves. Plant Sci. 2017, 256, 65–71. [Google Scholar] [CrossRef]
- Li, P.; Cheng, L.; Gao, H.; Jiang, C.; Tao, P. Heterogeneous behavior of PSII in soybean (Glycine max) leaves with identical PSII photochemistry efficiency under different high temperature treatments. J. Plant Physiol. 2009, 166, 1607–1615. [Google Scholar] [CrossRef]
- Saidi, Y.; Finka, A.; Muriset, M.; Bromberg, Z.; Weiss, Y.G.; Maathuis, F.J.M.; Goloubinoff, P. The heat shock response in moss plants is regulated by specific calcium-permeable channels in the plasma membrane. Plant Cell 2009, 21, 2829–2843. [Google Scholar] [CrossRef]
- Lima, R.; Santos, T.; Vieira, L.; Ferrarese, M.; Ferrarese-Filho, O.; Donatti, L.; Boeger, M.; Petkowicz, C. Heat stress causes alterations in the cell-wall polymers and anatomy of coffee leaves (Coffea arabica L.). Carbohyd. Polym. 2013, 93, 135–143. [Google Scholar] [CrossRef]
- Takahashi, S.; Whitney, S.; Itoh, S.; Maruyama, T.; Badger, M. Heat stress causes inhibition of the de novo synthesis of antenna proteins and photobleaching in cultured Symbiodinium. Proc. Natl. Acad. Sci. USA 2008, 105, 4203–4208. [Google Scholar] [CrossRef] [PubMed]
- Waszczak, C.; Carmody, M.; Kangasjärvi, J. Reactive oxygen species in plant signaling. Annu. Rev. Plant Biol. 2018, 69, 209–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, H.; Zhou, J.M. Malate circulation: Linking chloroplast metabolism to mitochondrial ROS. Trends Plant Sci. 2020, 25, 446–454. [Google Scholar] [CrossRef]
- Selinski, J.; Scheibe, R. Malate valves: Old shuttles with new perspectives. Plant Biol. 2019, 21, 21–30. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Xiong, Y.; Shi, J.; Chen, C.; Pan, Y.; Xue, T.; Xue, J.; Duan, Y. Stearic acid desaturase gene negatively regulates the thermotolerance of Pinellia ternata by modifying the saturated levels of fatty acids. Ind. Crop. Prod. 2021, 166, 113490. [Google Scholar] [CrossRef]
- Baird, C.H.; Niederlechner, S.; Beck, R.; Kallweit, A.R.; Wischmeyer, P.E. L-Threonine induces heat shock protein expression and decreases apoptosis in heat-stressed intestinal epithelial cells. Nutrition 2013, 29, 1404–1411. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Bio. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Ullrich, T.C.; Blaesse, M.; Huber, R. Crystal structure of ATP sulfurylase from Saccharomyces cerevisiae, a key enzyme in sulfate activation. EMBO J. 2001, 20, 316–329. [Google Scholar] [CrossRef]
- Yoshida, S.; Forno, D.A.; Cock, J.; Gomez, K.A. Laboratory Manual for Physiological Studies of Rice; IRRI: Las Banos, Philippines, 1976. [Google Scholar]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-the DESeq Package; EMBL: Heidelberg, Germany, 2012. [Google Scholar]
- Zou, S.C.; Wu, J.C.; Shahid, M.Q.; He, Y.H.; Lin, S.Q.; Liu, Z.H.; Yang, X.G. Identification of key taste components in loquat using widely targeted metabolomics. Food Chem. 2020, 323, 126822. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Yang, C.B.; Yuan, H.J.; Wang, Y.L.; Jabu, D.Z.; Xu, Q.J. Insights into the metabolic responses of two contrasting Tibetan hulless barley genotypes under low nitro-gen stress. Bioinformation 2019, 15, 845–851. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total | Upregulated | Downregulated |
|---|---|---|---|
| DY80 vs. DY80-CK | 355 | 262 | 93 |
| R974 vs. R974-CK | 82 | 63 | 19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Yu, H.; Huang, Z.; Yang, P.; Li, H.; Huang, G.; Tang, L.; Zhong, Z.; Hu, G.; Yu, G.; et al. Combined Analysis of Transcriptome and Metabolome Reveals the Heat Stress Resistance of Dongxiang Wild Rice at Seedling Stage. Plants 2025, 14, 1192. https://doi.org/10.3390/plants14081192
Zhang P, Yu H, Huang Z, Yang P, Li H, Huang G, Tang L, Zhong Z, Hu G, Yu G, et al. Combined Analysis of Transcriptome and Metabolome Reveals the Heat Stress Resistance of Dongxiang Wild Rice at Seedling Stage. Plants. 2025; 14(8):1192. https://doi.org/10.3390/plants14081192
Chicago/Turabian StyleZhang, Peng, Haipeng Yu, Zengying Huang, Pengfei Yang, Huijuan Li, Guanrong Huang, Lu Tang, Zhengzheng Zhong, Guocheng Hu, Guoping Yu, and et al. 2025. "Combined Analysis of Transcriptome and Metabolome Reveals the Heat Stress Resistance of Dongxiang Wild Rice at Seedling Stage" Plants 14, no. 8: 1192. https://doi.org/10.3390/plants14081192
APA StyleZhang, P., Yu, H., Huang, Z., Yang, P., Li, H., Huang, G., Tang, L., Zhong, Z., Hu, G., Yu, G., & Tong, H. (2025). Combined Analysis of Transcriptome and Metabolome Reveals the Heat Stress Resistance of Dongxiang Wild Rice at Seedling Stage. Plants, 14(8), 1192. https://doi.org/10.3390/plants14081192