
Weighted Gene Co-Expression Network Analysis Uncovers Core Drought Responsive Genes in Pecan (Carya illinoinensis)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
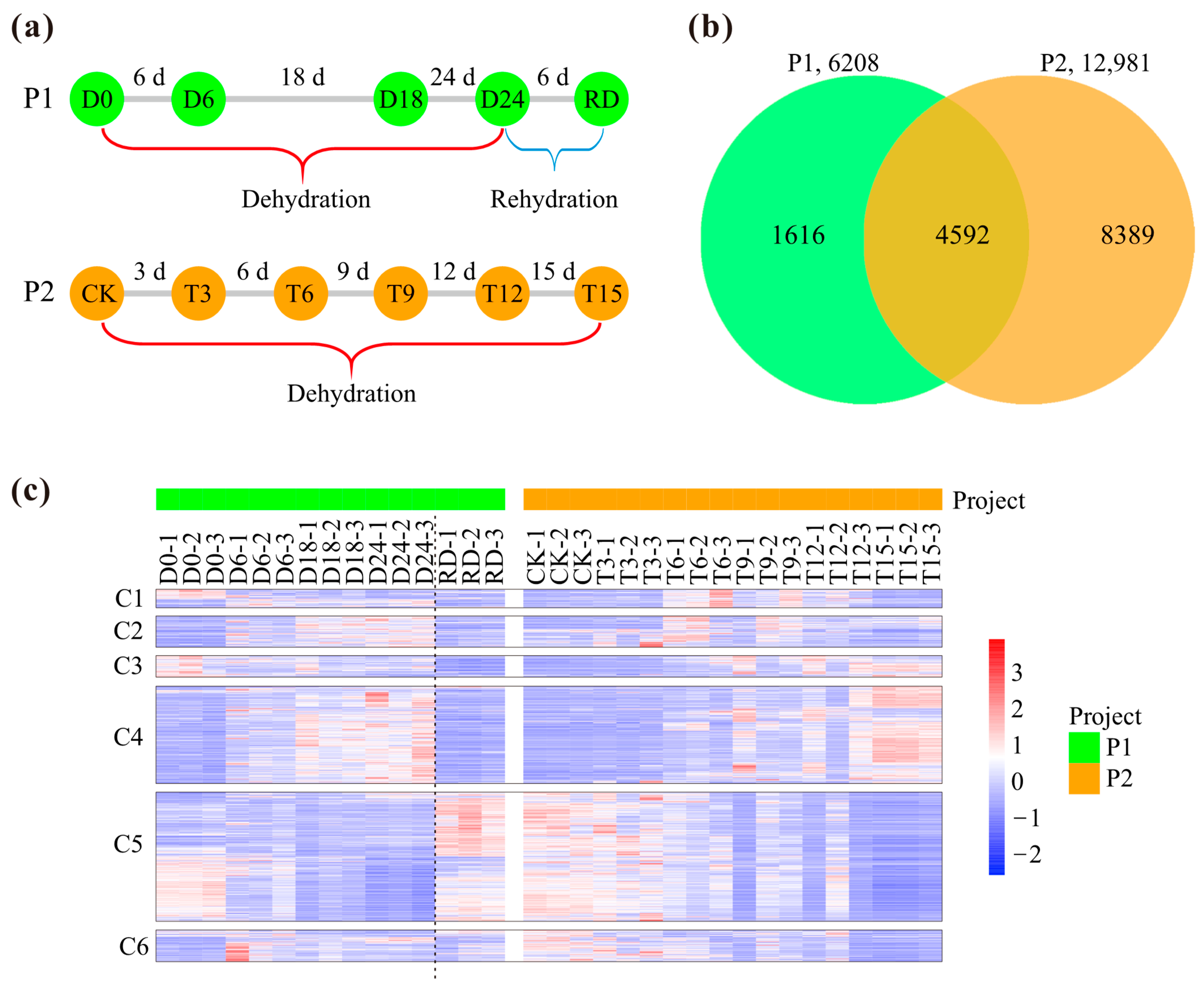
2.1. DEGs for Pecan in Response to Drought Stress
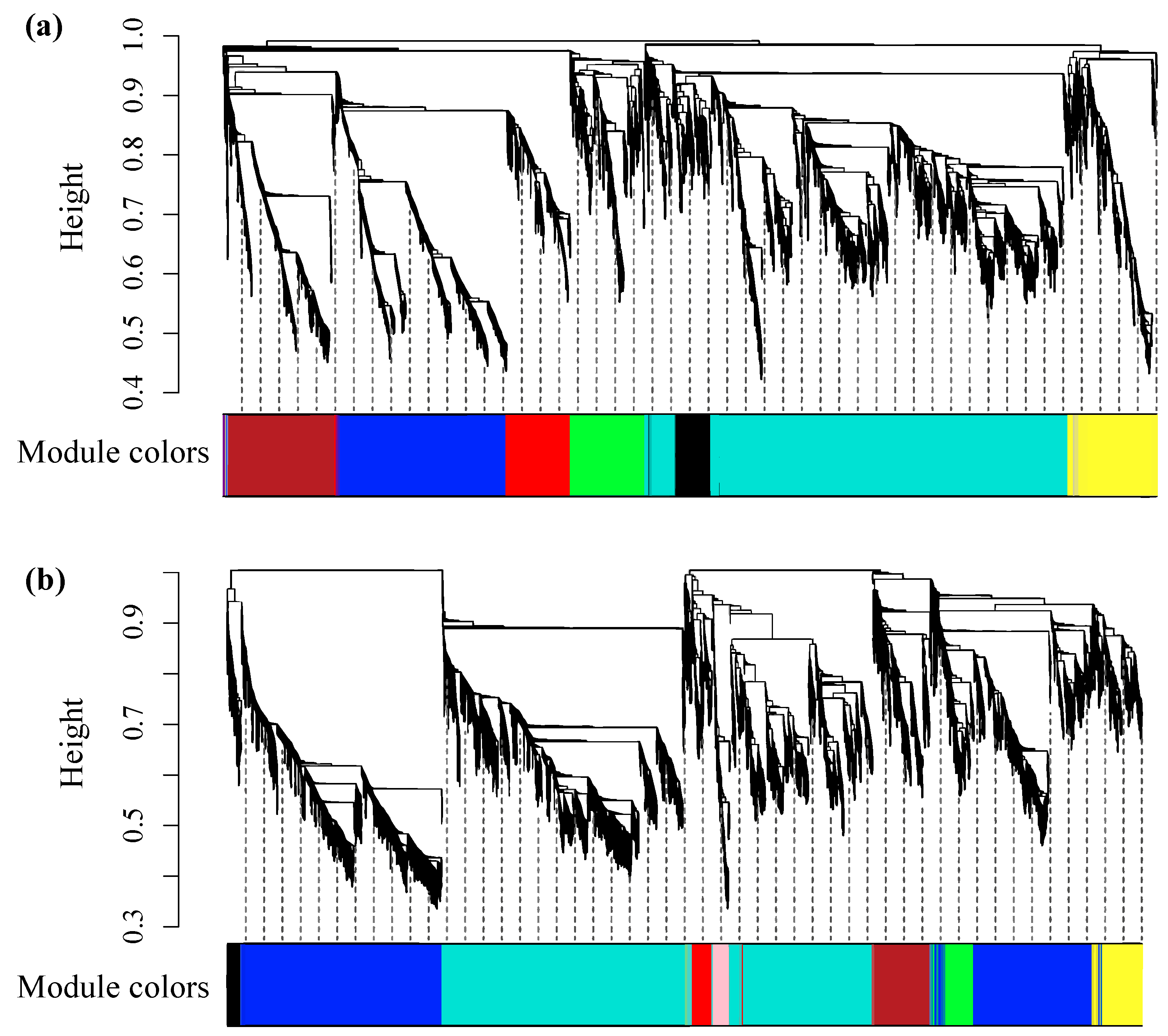
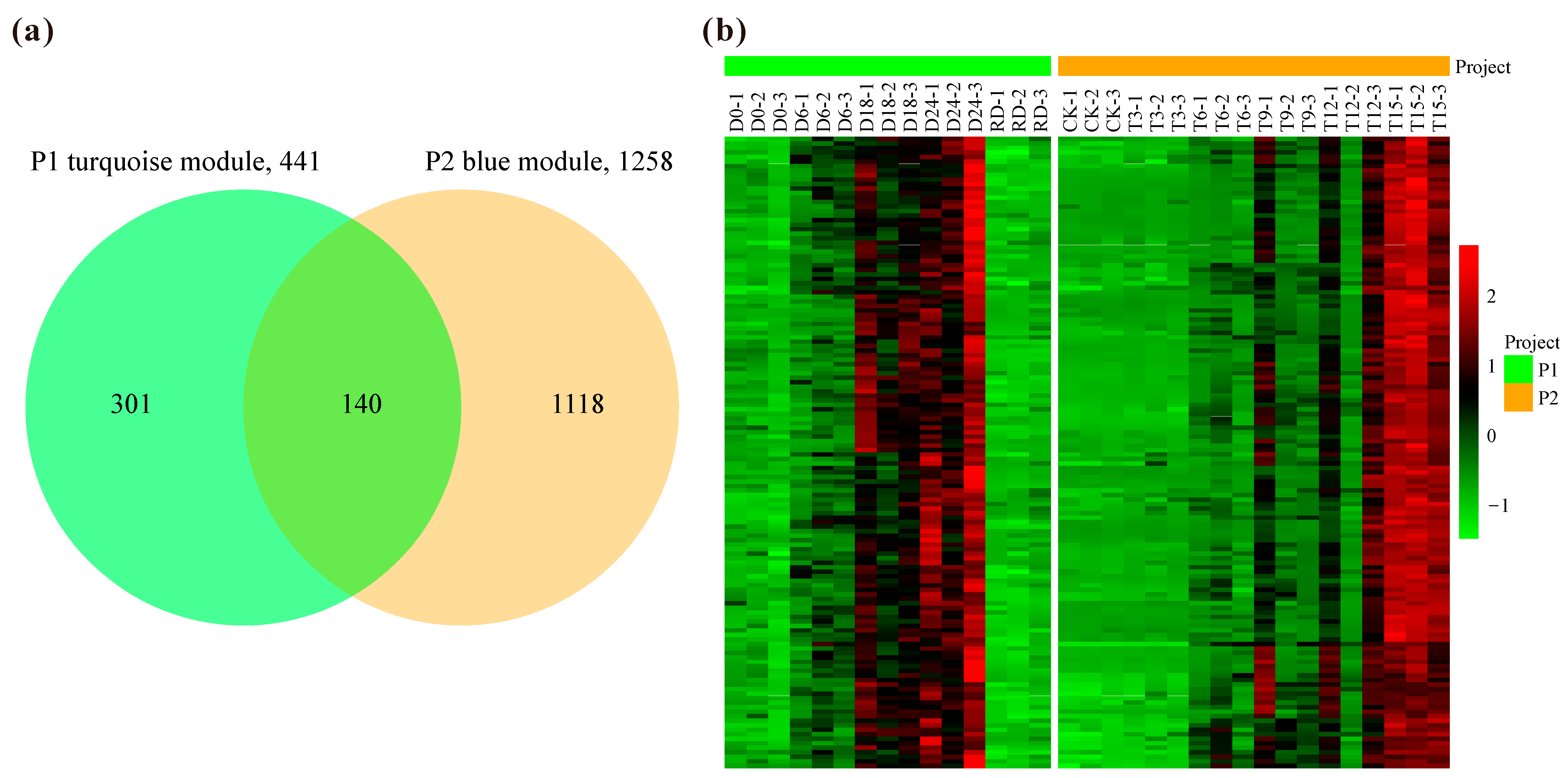
2.2. Co-Expression Networks Based on Drought-Related RNA-Seq
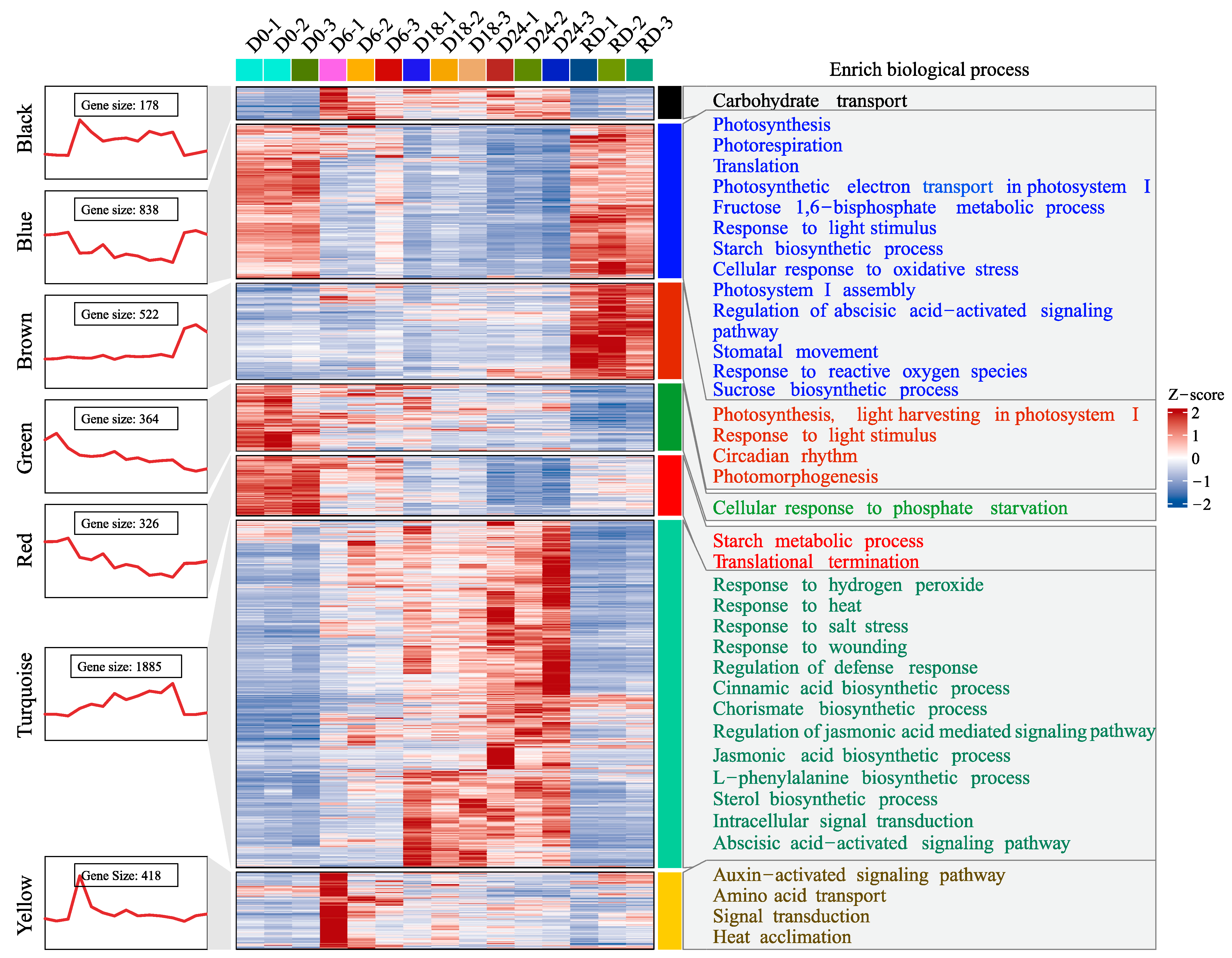
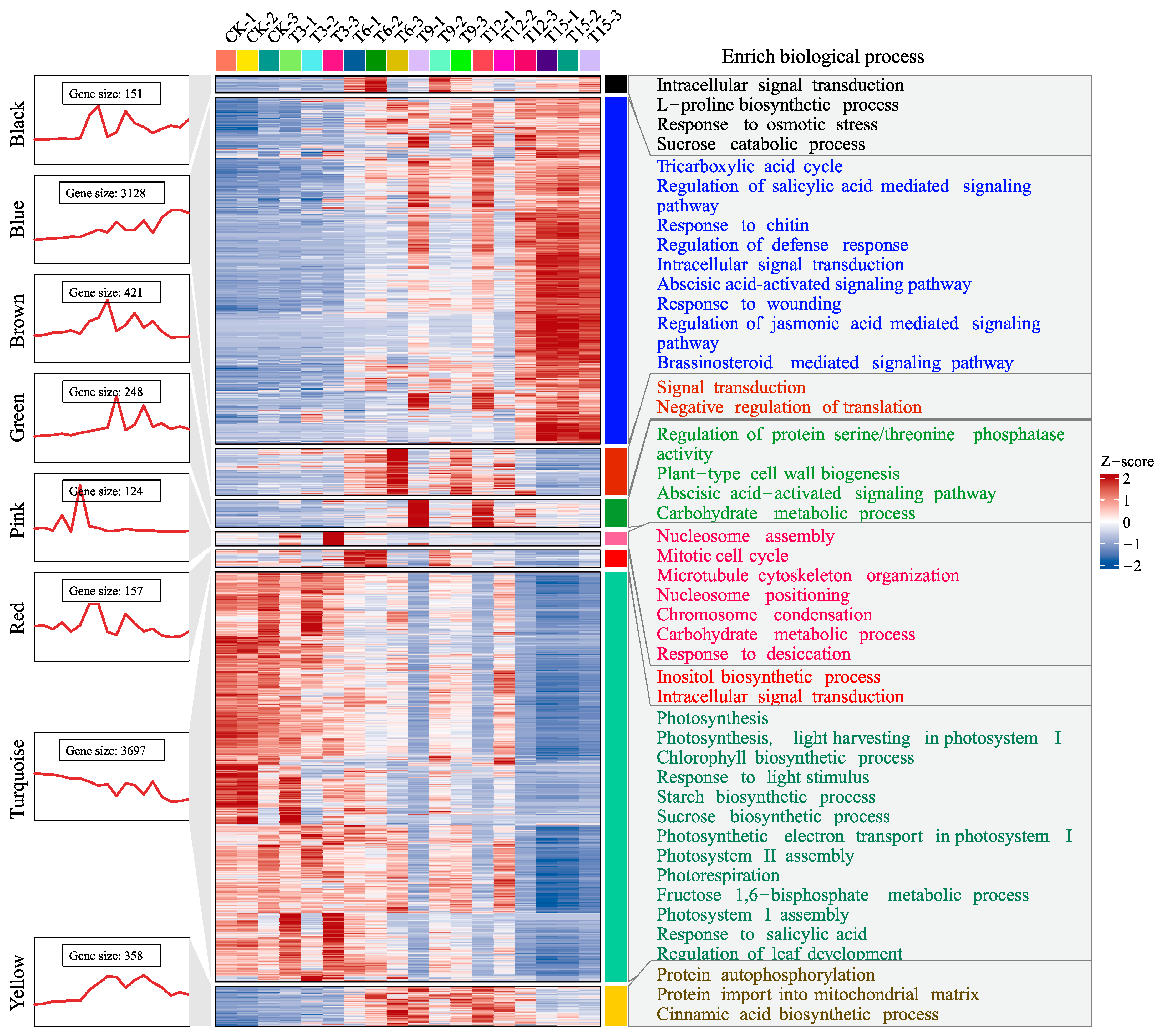
2.3. Expression Profile and Functional Enrichment for the Drought-Related Modules
2.4. Potential Core Drought-Responsive Genes
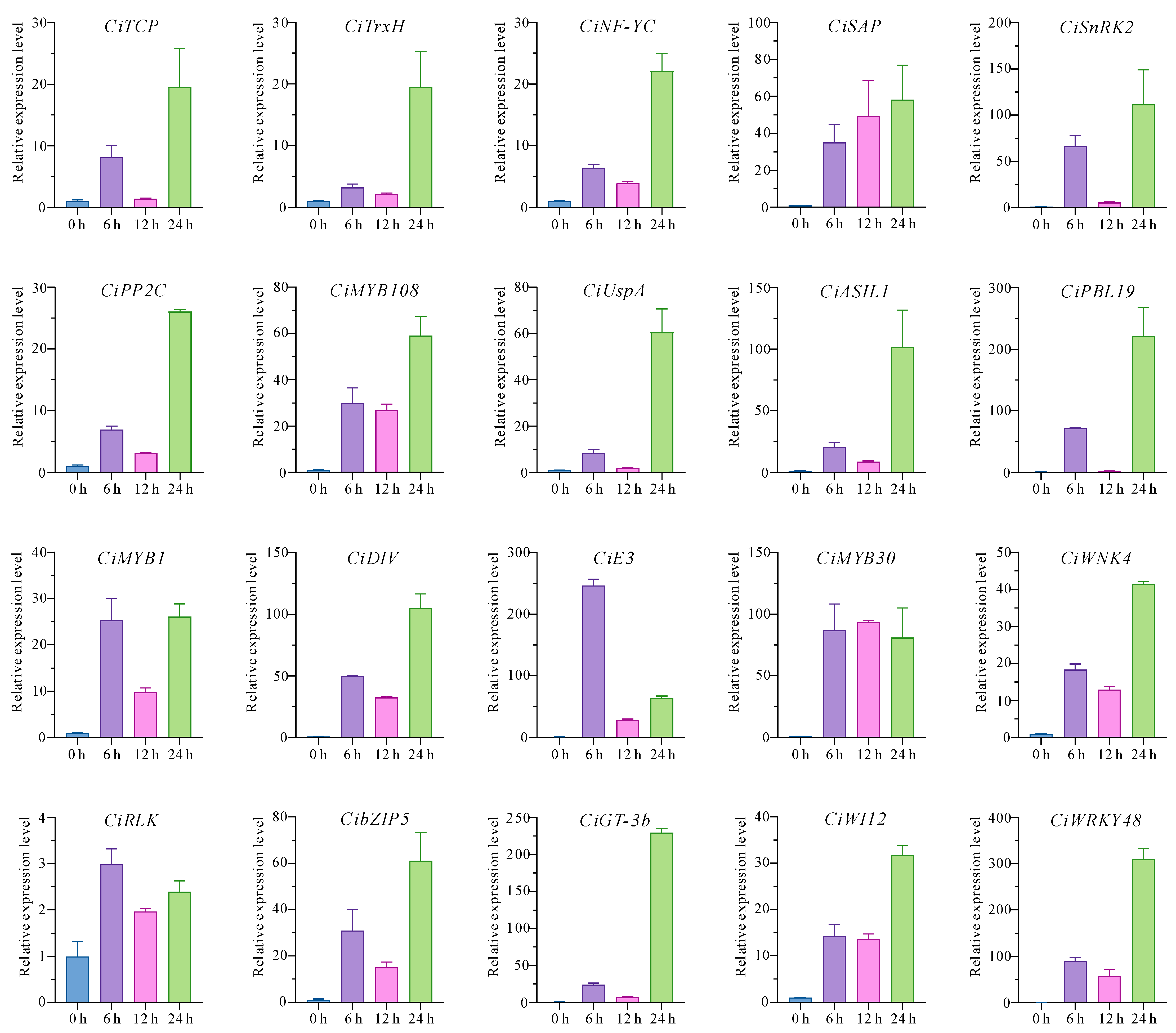
2.5. Validation of the Core Drought-Responsive Genes via RT-qPCR
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Data Processing
4.2. Identification of Differentially Expressed Genes (DEGs)
4.3. Construction of Co-Expression Network
4.4. Functional Enrichment Analysis of Modules
4.5. Selection of Hub Genes
4.6. Drought Stress Treatment
4.7. Validation the Responsiveness of Drought Related Genes via RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| WGCNA | Weighted Gene Co-expression Network Analysis |
| DEG | Differentially Expressed Gene |
| GO | Gene Ontology |
References
- Sallam, A.; Alqudah, A.M.; Dawood, M.F.; Baenziger, P.S.; Börner, A. Drought stress tolerance in wheat and barley: Advances in physiology, breeding and genetics research. Int. J. Mol. Sci. 2019, 20, 3137. [Google Scholar] [CrossRef] [PubMed]
- Cornic, G. Drought stress inhibits photosynthesis by decreasing stomatal aperture–not by affecting ATP synthesis. Trends Plant Sci. 2000, 5, 187–188. [Google Scholar] [CrossRef]
- Fracasso, A.; Trindade, L.M.; Amaducci, S. Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 2016, 16, 115. [Google Scholar] [CrossRef]
- Fang, Y.; Xiong, L. General mechanisms of drought response and their application in drought resistance improvement in plants. Cell. Mol. Life Sci. 2015, 72, 673–689. [Google Scholar] [CrossRef]
- Haghpanah, M.; Hashemipetroudi, S.; Arzani, A.; Araniti, F. Drought tolerance in plants: Physiological and molecular responses. Plants 2024, 13, 2962. [Google Scholar] [CrossRef]
- Yang, X.; Lu, M.; Wang, Y.; Wang, Y.; Liu, Z.; Chen, S. Response mechanism of plants to drought stress. Horticulturae 2021, 7, 50. [Google Scholar] [CrossRef]
- Song, Y.; Li, F.; Ali, M.; Li, X.; Zhang, X.; Ahmed, Z.F. Advances in protein kinase regulation of stress responses in fruits and vegetables. Int. J. Mol. Sci. 2025, 26, 768. [Google Scholar] [CrossRef] [PubMed]
- González-Mendoza, V.M.; Sánchez-Sandoval, M.; Castro-Concha, L.A.; Hernández-Sotomayor, S.T. Phospholipases C and D and their role in biotic and abiotic stresses. Plants 2021, 10, 921. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.; Li, L.; Reynolds, M.; Mao, X.; Jing, R. Exploitation of drought tolerance-related genes for crop improvement. Int. J. Mol. Sci. 2021, 22, 10265. [Google Scholar] [CrossRef]
- Thompson, T.E.; Conner, P.J. Pecan. In Fruit Breeding; Springer: Berlin/Heidelberg, Germany, 2012; pp. 771–801. [Google Scholar]
- Bilharva, M.G.; Martins, C.R.; Hamann, J.J.; Fronza, D.; De Marco, R.; Malgarim, M.B. Pecan: From research to the Brazilian reality. J. Exp. Agric. Int. 2018, 23, 1–16. [Google Scholar] [CrossRef]
- Fronza, D.; Hamann, J.J.; Both, V.; Anese, R.d.O.; Meyer, E.A. Pecan cultivation: General aspects. Ciência Rural 2018, 48, e20170179. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Sabharanjak, S.M.; Zengin, G.; Mollica, A.; Szostak, A.; Simirgiotis, M.; Huminiecki, Ł.; Horbanczuk, O.K.; Nabavi, S.M.; Mocan, A. Pecan nuts: A review of reported bioactivities and health effects. Trends Food Sci. Tech. 2018, 71, 246–257. [Google Scholar] [CrossRef]
- Xu, Q.; He, H.; He, B.; Li, T.; Liu, Y.; Zhu, S.; Zhang, G. Nitrogen allocation tradeoffs within-leaf between photosynthesis and high-temperature adaptation among different varieties of pecan (Carya illinoinensis [Wangenh.] K. Koch). Plants 2022, 11, 2828. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Chen, Y.; Lou, W.; Jia, X.; Zhai, M.; Xuan, J.; Guo, Z.; Li, Y. Identification of suitable reference genes for normalization of real-time quantitative PCR data in pecan (Carya illinoinensis). Trees 2020, 34, 1233–1241. [Google Scholar] [CrossRef]
- Kaur, A.; Ferguson, L.; Maness, N.; Carroll, B.; Reid, W.; Zhang, L. Spring freeze damage of pecan bloom: A review. Horticulturae 2020, 6, 82. [Google Scholar] [CrossRef]
- Yang, B.; Yang, C.; Chen, J.; Ren, H.; Wang, K.; Liu, L.; Yao, X. CiNAC2 positively regulates drought stress tolerance by promoting superoxide dismutase activity in pecan (Carya illinoinensis). Hortic. Plant J. 2025, 11, 133–144. [Google Scholar] [CrossRef]
- Othman, Y.; Steele, C.; VanLeeuwen, D.; Heerema, R.; Bawazir, S.; St. Hilaire, R. Remote sensing used to detect moisture status of pecan orchards grown in a desert environment. Int. J. Remote Sens. 2014, 35, 949–966. [Google Scholar] [CrossRef]
- Wang, H.; He, T.; Huang, C.; Wang, K.; Shi, D.; Si, X.; Xu, Y.; Lyu, S.; Huang, J.; Li, Y. Genome-wide identification of KCS gene family in Carya illinoinensis and their roles under abiotic stress conditions. Sci. Hortic. 2023, 321, 112343. [Google Scholar] [CrossRef]
- Garrot, D., Jr.; Kilby, M.W.; Fangmeier, D.D.; Husman, S.H.; Ralowicz, A.E. Production, growth and nut quality in pecans under water stress based on the crop water stress index. J. Am. Soc. Hort. Sci. 1993, 118, 694–698. [Google Scholar] [CrossRef]
- Clermont, K.; Graham, C.J.; Lloyd, S.W.; Grimm, C.C.; Randall, J.J.; Mattison, C.P. Proteomic analysis of pecan (Carya illinoinensis) nut development. Foods 2023, 12, 866. [Google Scholar] [CrossRef]
- Stein, L.A.; McEachern, G.R.; Storey, J.B. Summer and fall moisture stress and irrigation scheduling influence pecan growth and production. HortScience 1989, 24, 607–611. [Google Scholar] [CrossRef]
- ShalekBriski, A.; Brorsen, B.W.; Biermacher, J.T.; Rohla, C.T.; Chaney, W. Effect of irrigation method on tree growth, foliar nutrient levels, and nut characteristics of young pecan trees in the southern great plains. HortTechnology 2019, 29, 109–113. [Google Scholar] [CrossRef]
- Zhu, K.; Wei, L.; Ma, W.; Hu, X.; Zhao, J.; Tan, P.; Liu, H.; Feng, G.; Fan, P.; Peng, F. Transcriptome analysis of pecan (Carya illinoinensis) differentially expressed genes in response to drought stress. Forests 2023, 14, 608. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, L.; Lai, J.; Zhao, H.; Song, W. Effects of drought stress and water recovery on physiological responses and gene expression in maize seedlings. BMC Plant Biol. 2018, 18, 68. [Google Scholar] [CrossRef]
- Shi, D.; Wang, J.; Bai, Y.; Liu, Y. Transcriptome sequencing of okra (Abelmoschus esculentus L. Moench) uncovers differently expressed genes responding to drought stress. J. Plant Biochem. Biot. 2020, 29, 155–170. [Google Scholar] [CrossRef]
- Mo, Z.; Jiang, X.; Zhang, Y.; Zhai, M.; Hu, L.; Xuan, J. Weighted gene co-expression network analysis reveals key pathways and hub genes associated with successful grafting in pecan (Carya illinoinensis). Forests 2023, 14, 835. [Google Scholar] [CrossRef]
- Shang, P.; Bi, L.; Li, W.; Zhou, X.; Feng, Y.; Wu, J.; Zeng, B. Exploration of key genes and pathways in response to submergence stress in red clover (Trifolium pratense L.) by WGCNA. BMC Plant Biol. 2025, 25, 4. [Google Scholar] [CrossRef]
- Mo, Z.; Zhang, Y.; Hou, M.; Hu, L.; Zhai, M.; Xuan, J. Transcriptional dynamics reveals the asymmetrical events underlying graft union formation in pecan (Carya illinoinensis). Tree Physiol. 2024, 44, tpae040. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, M.; Gu, W.; Chen, Z.; Gu, Y.; Pei, L.; Tian, R. Effect of drought on photosynthesis, total antioxidant capacity, bioactive component accumulation, and the transcriptome of Atractylodes lancea. BMC Plant Biol. 2021, 21, 293. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, W.; Wang, B.; Yang, L. Molecular and physiological responses of two quinoa genotypes to drought stress. Front. Genet. 2024, 15, 1439046. [Google Scholar] [CrossRef] [PubMed]
- Assmann, S.M.; Jegla, T. Guard cell sensory systems: Recent insights on stomatal responses to light, abscisic acid, and CO2. Curr. Opin. Plant Biol. 2016, 33, 157–167. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, W.; Ma, J.; Cheng, Z.; Zhang, X.; Liu, X.; Wu, X.; Zhang, J. An integrated framework for drought stress in plants. Int. J. Mol. Sci. 2024, 25, 9347. [Google Scholar] [CrossRef]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Cruz de Carvalho, M.H. Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sarwat, M.; Tuteja, N. Hormonal signaling to control stomatal movement during drought stress. Plant Gene 2017, 11, 143–153. [Google Scholar] [CrossRef]
- Daszkowska-Golec, A.; Szarejko, I. Open or close the gate–stomata action under the control of phytohormones in drought stress conditions. Front. Plant Sci. 2013, 4, 138. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, J.; Guo, Z.; Lu, S.; He, S.; Shu, W.; Zhou, B. Increased abscisic acid levels in transgenic tobacco over-expressing 9 cis-epoxycarotenoid dioxygenase influence H2O2 and NO production and antioxidant defences. Plant Cell Environ. 2009, 32, 509–519. [Google Scholar] [CrossRef]
- Dombrecht, B.; Xue, G.P.; Sprague, S.J.; Kirkegaard, J.A.; Ross, J.J.; Reid, J.B.; Fitt, G.P.; Sewelam, N.; Schenk, P.M.; Manners, J.M. MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 2007, 19, 2225–2245. [Google Scholar] [CrossRef]
- Savchenko, T.V.; Rolletschek, H.; Dehesh, K. Jasmonates-mediated rewiring of central metabolism regulates adaptive responses. Plant Cell Physiol. 2019, 60, 2613–2620. [Google Scholar] [CrossRef]
- Anjum, S.; Wang, L.; Farooq, M.; Khan, I.; Xue, L. Methyl jasmonate-induced alteration in lipid peroxidation, antioxidative defence system and yield in soybean under drought. J. Agron. Crop Sci. 2011, 197, 296–301. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhu, H.; Zhu, H.; Tao, Y.; Liu, C.; Liu, J.; Yang, F.; Li, M. Exogenous ABA enhances the antioxidant defense system of maize by regulating the AsA-GSH cycle under drought stress. Sustainability 2022, 14, 3071. [Google Scholar] [CrossRef]
- Savchenko, T.; Kolla, V.A.; Wang, C.-Q.; Nasafi, Z.; Hicks, D.R.; Phadungchob, B.; Chehab, W.E.; Brandizzi, F.; Froehlich, J.; Dehesh, K. Functional convergence of oxylipin and abscisic acid pathways controls stomatal closure in response to drought. Plant Physiol. 2014, 164, 1151–1160. [Google Scholar] [CrossRef]
- Buti, M.; Baldoni, E.; Formentin, E.; Milc, J.; Frugis, G.; Lo Schiavo, F.; Genga, A.; Francia, E. A meta-analysis of comparative transcriptomic data reveals a set of key genes involved in the tolerance to abiotic stresses in rice. Int. J. Mol. Sci. 2019, 20, 5662. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lian, Y.; Gong, P.; Song, L.; Hu, J.; Pang, H.; Ren, Y.; Xin, Z.; Wang, Z.; Lin, T. Network of the transcriptome and metabolomics reveals a novel regulation of drought resistance during germination in wheat. Ann. Bot. 2022, 130, 717–735. [Google Scholar] [CrossRef]
- Zhou, Y.; Bai, Y.H.; Han, F.X.; Chen, X.; Wu, F.S.; Liu, Q.; Ma, W.Z.; Zhang, Y.Q. Transcriptome sequencing and metabolome analysis reveal the molecular mechanism of Salvia miltiorrhiza in response to drought stress. BMC Plant Biol. 2024, 24, 446. [Google Scholar] [CrossRef] [PubMed]
- Huang, B. Role of root morphological and physiological characteristics in drought resistance of plants. In Plant-Environment Interactions; Marcel Dekker Inc.: New York, NY, USA, 2000; pp. 39–64. [Google Scholar]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Li, S.; An, X.; Liu, X.; Qin, H.; Wang, D. Transgenic expression of MYB15 confers enhanced sensitivity to abscisic acid and improved drought tolerance in Arabidopsis thaliana. J. Genet. Genom. 2009, 36, 17–29. [Google Scholar] [CrossRef]
- Mengiste, T.; Chen, X.; Salmeron, J.; Dietrich, R. The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 2003, 15, 2551–2565. [Google Scholar] [CrossRef]
- Wu, Y.; Li, T.; Cheng, Z.; Zhao, D.; Tao, J. R2R3-MYB transcription factor PlMYB108 confers drought tolerance in herbaceous peony (Paeonia lactiflora pall.). Int. J. Mol. Sci. 2021, 22, 11884. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Mu, Y.; Guan, L.; Wu, F.; Liu, K.; Li, M.; Wang, N.; Zhuang, Z.; Zhao, Y. PwuWRKY48 confers drought tolerance in Populus wulianensis. Forests 2024, 15, 302. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Zhang, D.; Tang, X.; Li, Z.; Shen, C.; Han, X.; Deng, W.; Yin, W.; Xia, X. PtrWRKY75 overexpression reduces stomatal aperture and improves drought tolerance by salicylic acid-induced reactive oxygen species accumulation in poplar. Environ. Exp. Bot. 2020, 176, 104117. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujita, Y.; Sayama, H.; Kidokoro, S.; Maruyama, K.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 2010, 61, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Wei, J.; Huang, G.; Zhang, D.; Liu, Y.; Zhang, L. Homologous HAP5 subunit from Picea wilsonii improved tolerance to salt and decreased sensitivity to ABA in transformed Arabidopsis. Planta 2013, 238, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, J.; Guo, S.; Xie, X.; Yan, L.; Chen, H.; Zhang, H.; Bu, X.; Zheng, L.; Wang, Y. RtNAC055 promotes drought tolerance via a stomatal closure pathway linked to methyl jasmonate/hydrogen peroxide signaling in Reaumuria trigyna. Hortic. Res. 2024, 11, uhae001. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.T.; Bentley, N.B.; Bhattarai, G.; Jenkins, J.W.; Sreedasyam, A.; Alarcon, Y.; Bock, C.; Boston, L.B.; Carlson, J.; Cervantes, K. Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nat. Commun. 2021, 12, 4125. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Ge, Q.; Li, B.; Tong, Y.; Zhang, A.; Li, Z.; Kuang, T.; Lu, C. Tolerance of photosynthesis to photoinhibition, high temperature and drought stress in flag leaves of wheat: A comparison between a hybridization line and its parents grown under field conditions. Plant Sci. 2006, 171, 389–397. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, M.; Li, Y.; Xuan, J.; Zhang, Y.; Wang, T.; Zhai, M.; Wang, G.; Hu, L.; Mo, Z. Weighted Gene Co-Expression Network Analysis Uncovers Core Drought Responsive Genes in Pecan (Carya illinoinensis). Plants 2025, 14, 833. https://doi.org/10.3390/plants14060833
Hou M, Li Y, Xuan J, Zhang Y, Wang T, Zhai M, Wang G, Hu L, Mo Z. Weighted Gene Co-Expression Network Analysis Uncovers Core Drought Responsive Genes in Pecan (Carya illinoinensis). Plants. 2025; 14(6):833. https://doi.org/10.3390/plants14060833
Chicago/Turabian StyleHou, Mengxin, Yongrong Li, Jiping Xuan, Yan Zhang, Tao Wang, Min Zhai, Guoming Wang, Longjiao Hu, and Zhenghai Mo. 2025. "Weighted Gene Co-Expression Network Analysis Uncovers Core Drought Responsive Genes in Pecan (Carya illinoinensis)" Plants 14, no. 6: 833. https://doi.org/10.3390/plants14060833
APA StyleHou, M., Li, Y., Xuan, J., Zhang, Y., Wang, T., Zhai, M., Wang, G., Hu, L., & Mo, Z. (2025). Weighted Gene Co-Expression Network Analysis Uncovers Core Drought Responsive Genes in Pecan (Carya illinoinensis). Plants, 14(6), 833. https://doi.org/10.3390/plants14060833