
Integration of mRNA and miRNA Analysis Reveals the Regulation of Salt Stress Response in Rapeseed (Brassica napus L.)


 ,
,
Abstract
1. Introduction
2. Results
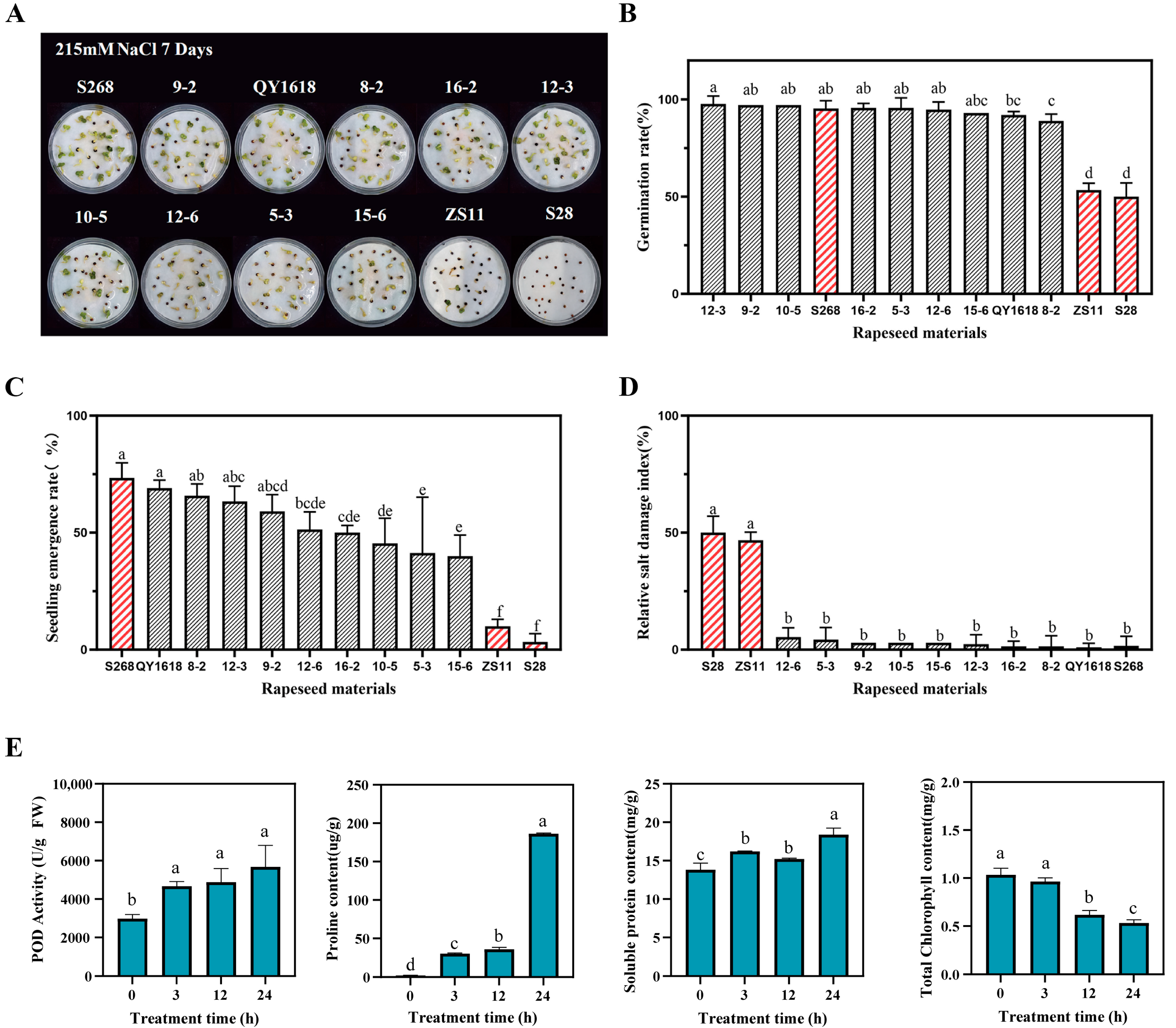
2.1. Evaluation of Salt Stress Tolerance in Different Rapeseed Cultivars
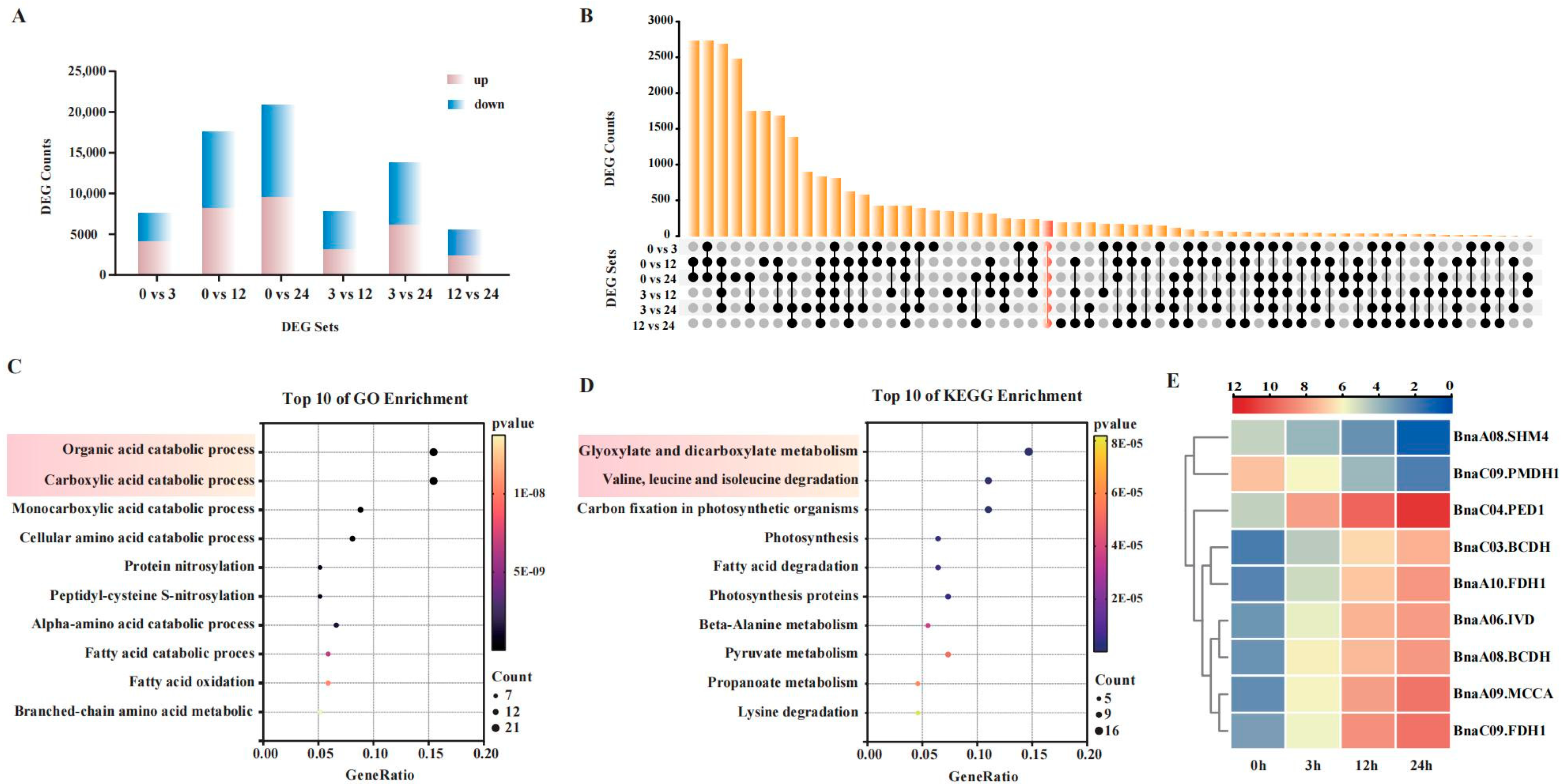
2.2. Functional Enrichment and Expression Dynamics of Core Genes in Rapeseed Cultivar S268 Under Salt Stress
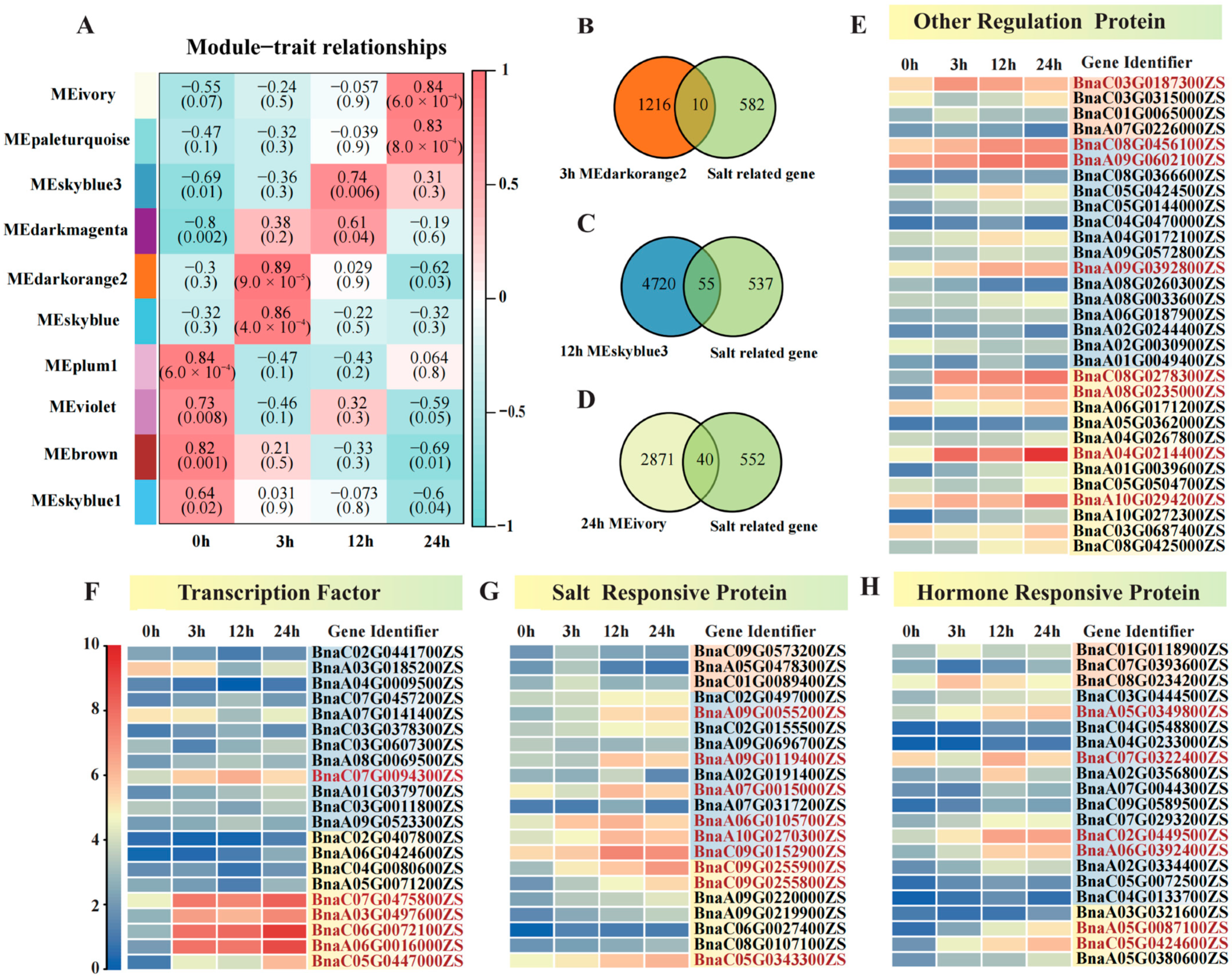
2.3. Co-Expression Network Analysis and Time Point-Specific Modules in Rapeseed S268 Under Salt Stress
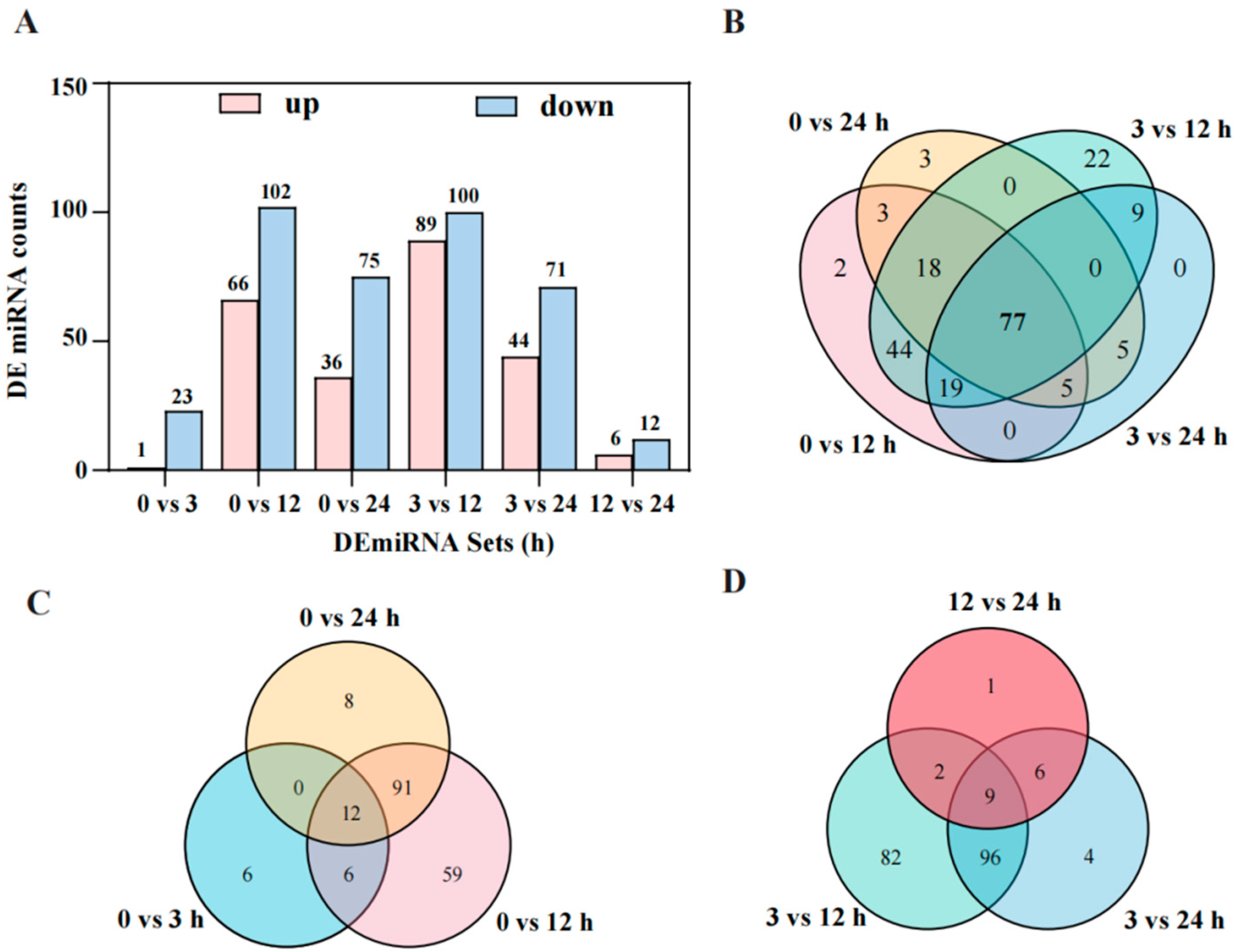
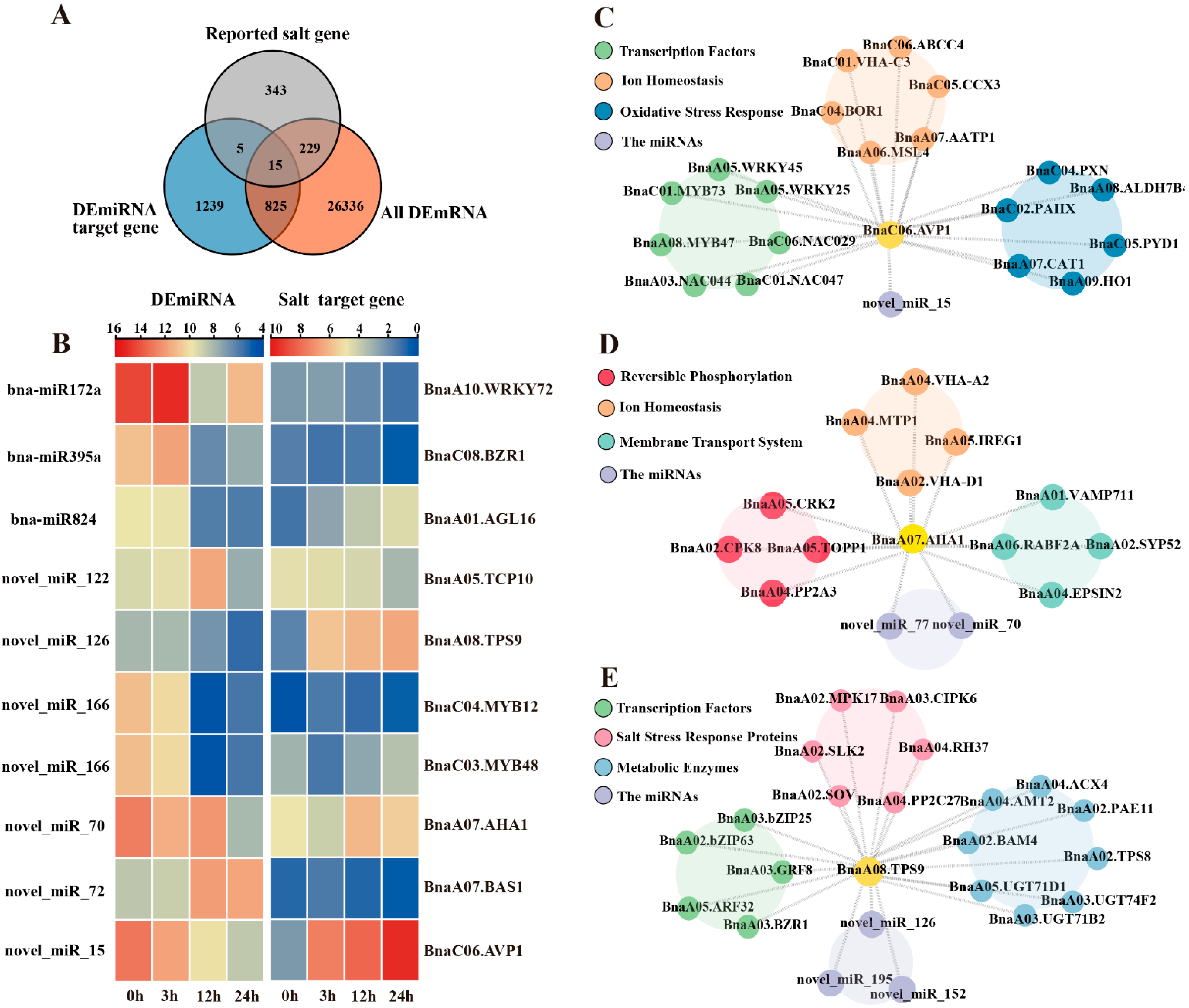
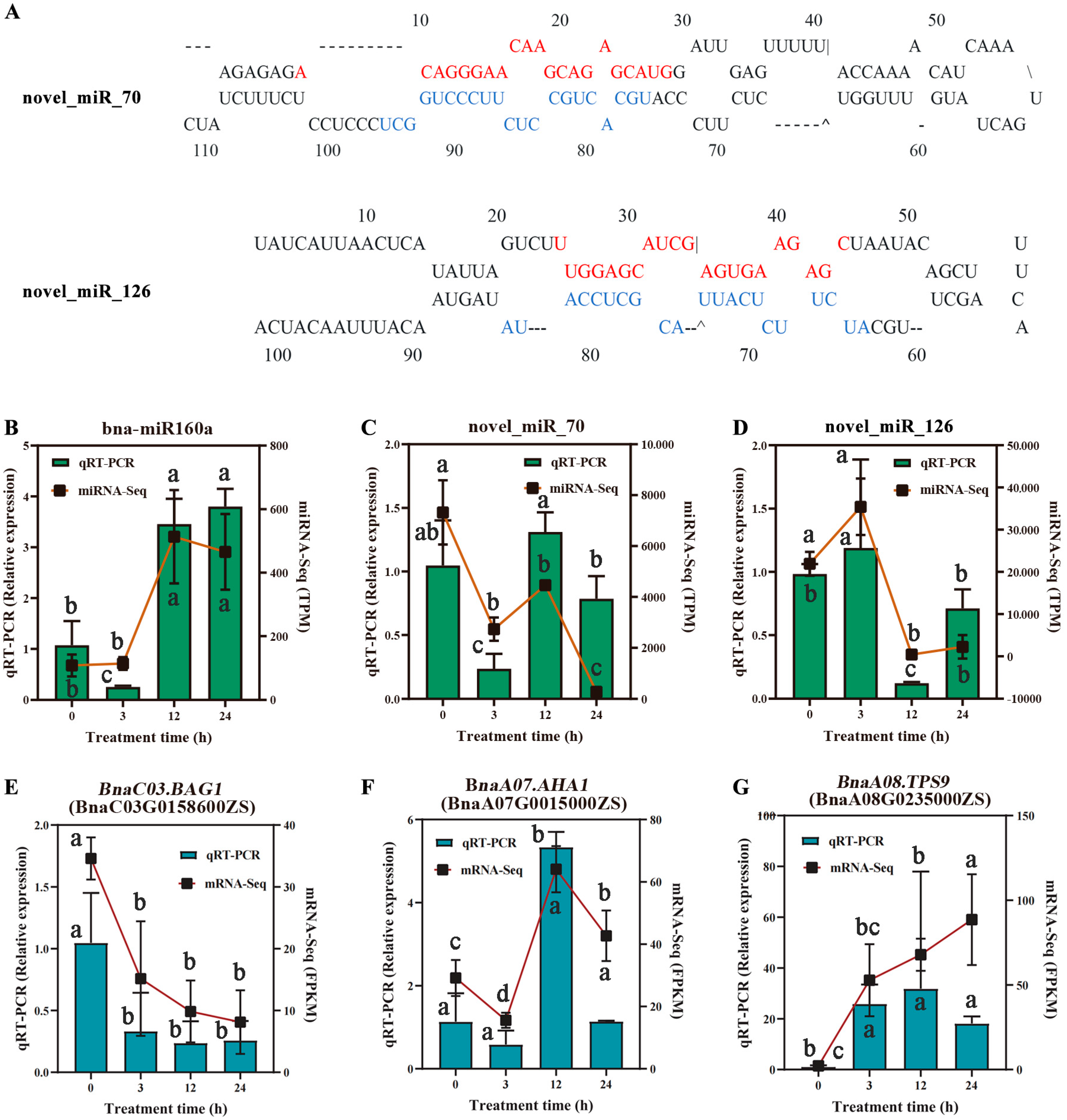
2.4. Identification of Differentially Expressed miRNAs (DEmiRNAs) and Their Target Genes Under Salt Stress
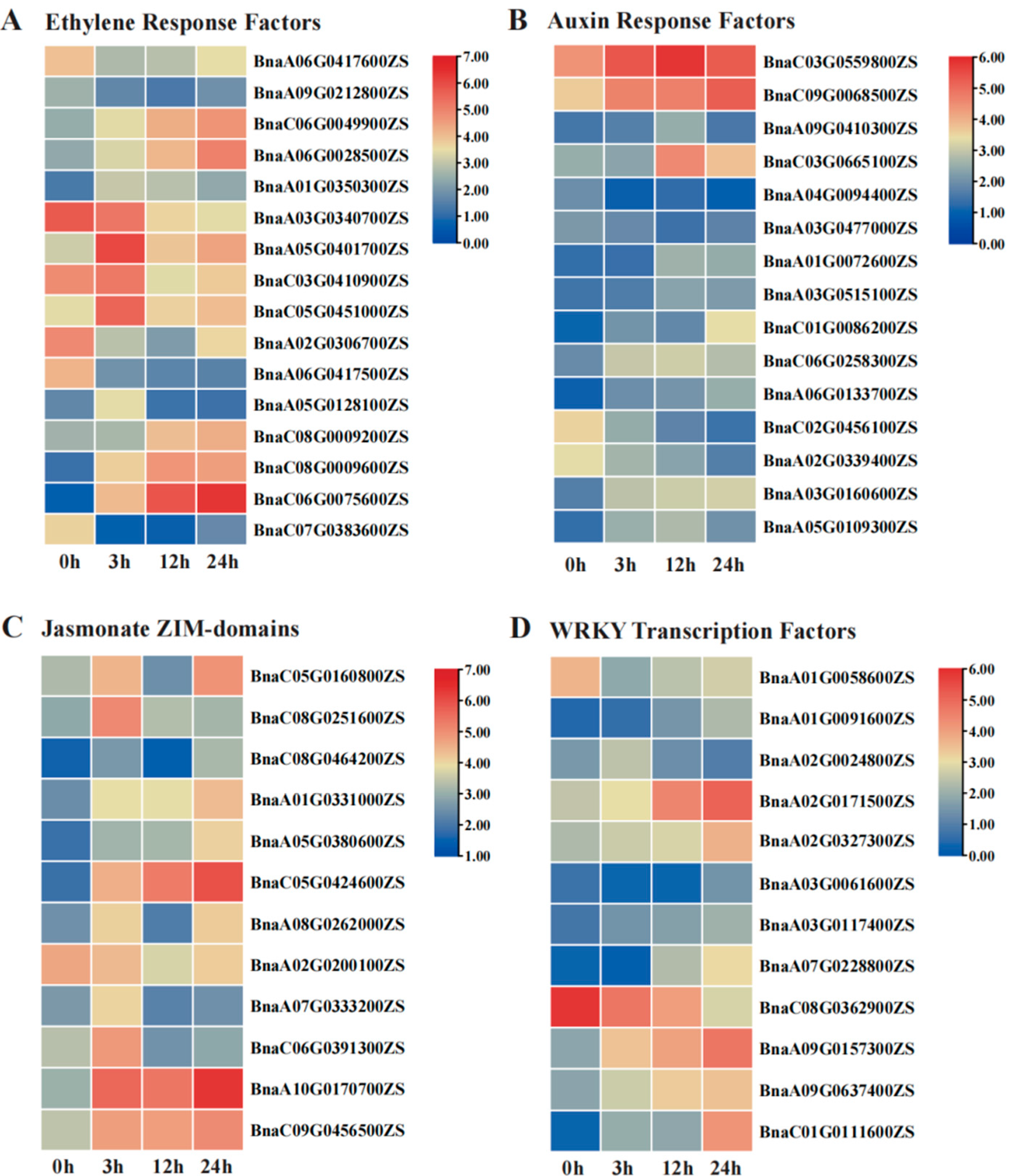
2.5. Functional Characterization of Salt-Responsive miRNA-mRNA Interactions Under Salt Stress
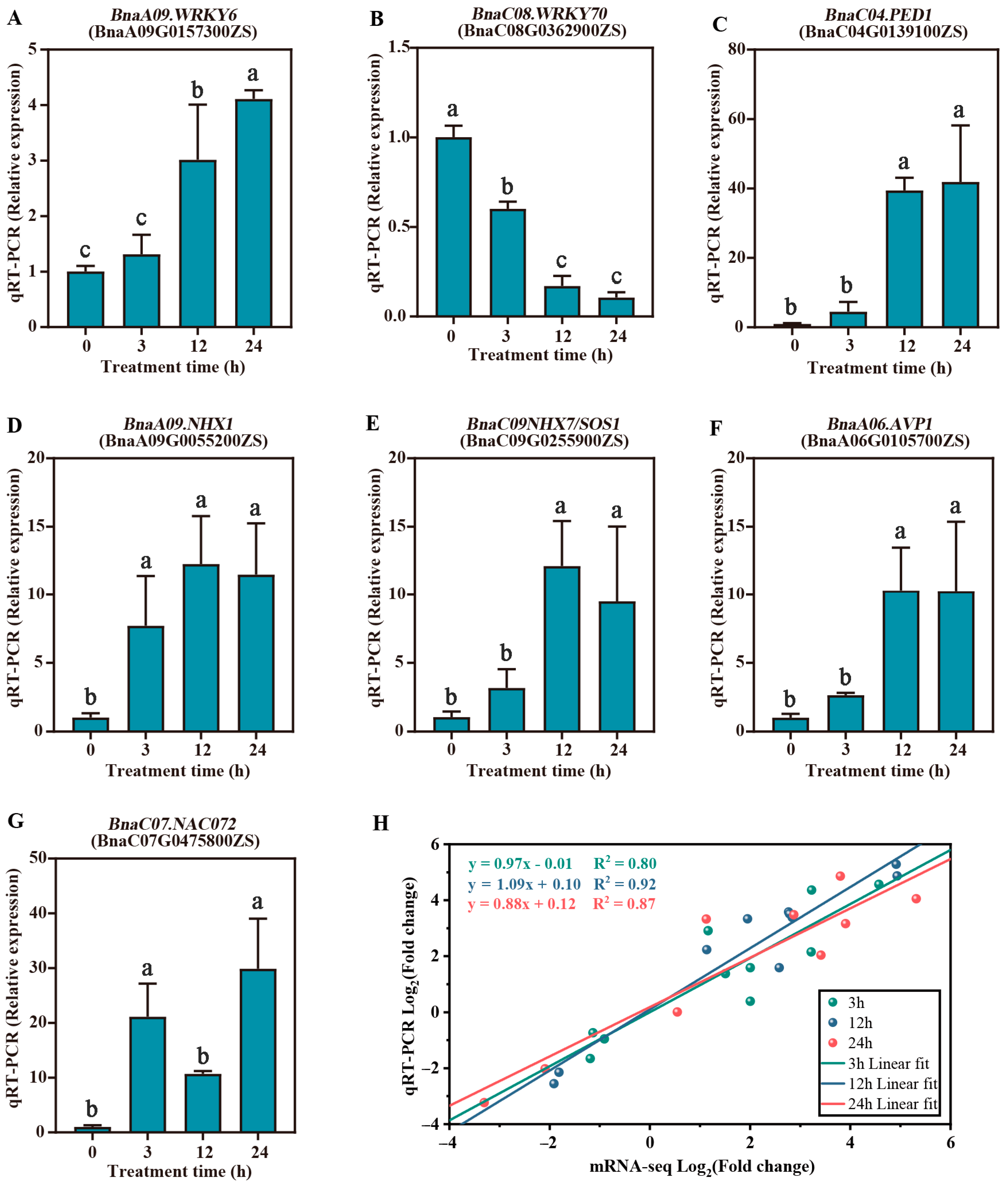
2.6. Validation of RNA-Seq and miRNA-Seq by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Stress Treatments
4.2. Determination of Physiological and Biochemical Indicators
4.3. Preparation of Plant Materials for mRNA-Seq and miRNA-Seq
4.4. Construction and Sequencing of mRNA-Seq and miRNA-Seq Libraries
4.5. miRNA Identification and Target Gene Prediction
4.6. Analysis of the Differential Expression of mRNA and miRNA
4.7. Co-Expression Network Construction
4.8. GO and KEGG Pathway Enrichment Analysis
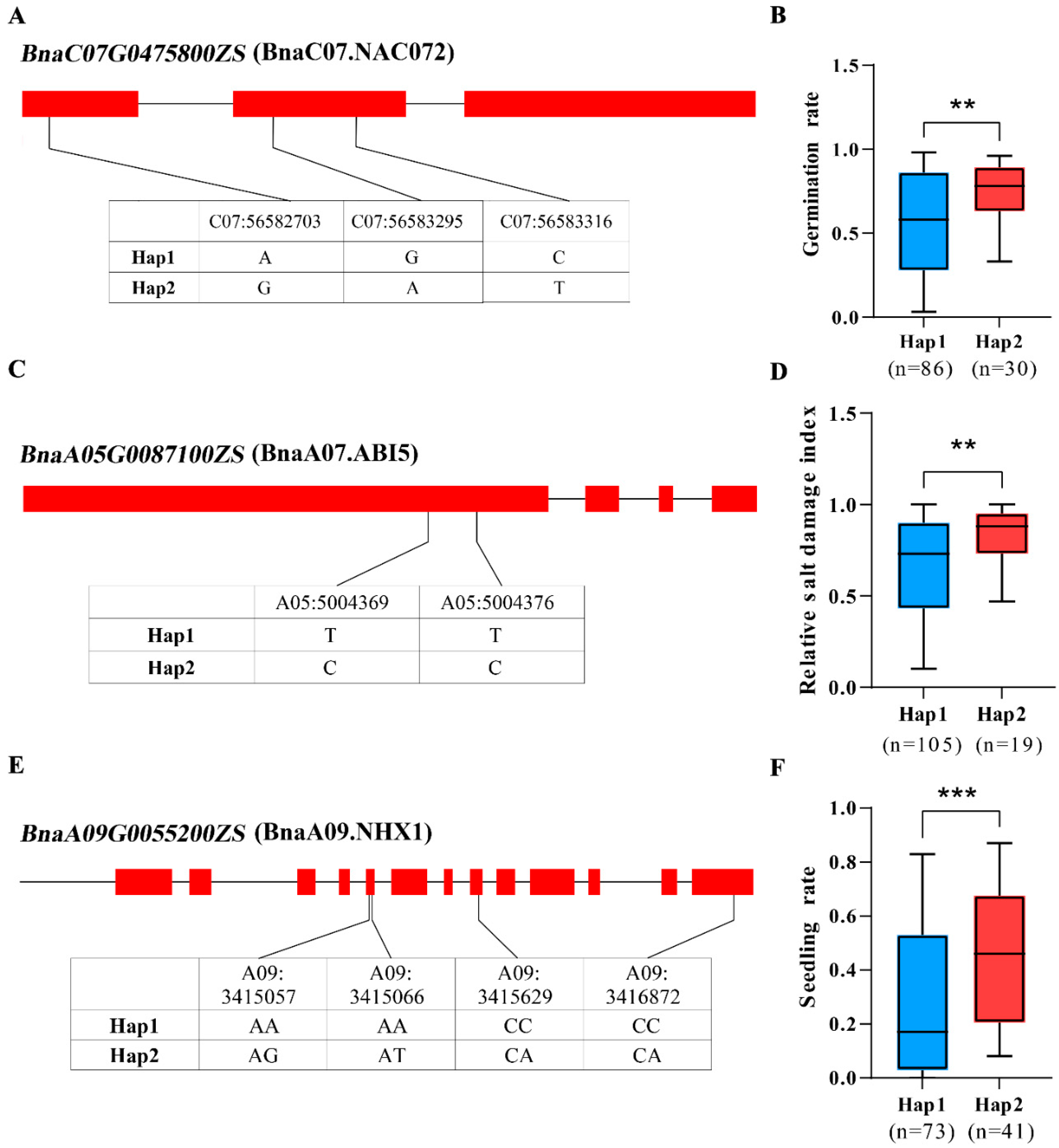
4.9. Haplotype Analysis
4.10. Validation of mRNA and miRNA Expressions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| BRs | Brassinosteroids |
| cDNA | Complementary DNA |
| DEGs | Differentially expressed genes |
| DEmiRNAs | Differentially expressed miRNAs |
| FDR | False discovery rate |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| JA | Jasmonic acid |
| RT-qPCR | Quantitative reverse transcription PCR |
| ROS | Reactive oxygen species |
| TPM | Transcripts per million |
| NGS | Next-generation sequencing |
| WGCNA | Weighted gene co-expression network analysis |
References
- Hopmans, J.W.; Qureshi, A.S.; Kisekka, I.; Munns, R.; Grattan, S.R.; Rengasamy, P.; Ben-Gal, A.; Assouline, S.; Javaux, M.; Minhas, P.S.; et al. Chapter One-Critical knowledge gaps and research priorities in global soil salinity. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: New York, NY, USA, 2021; pp. 1–191. ISBN 9780128245903. [Google Scholar]
- Jin, T.; Hao, X.S.; Huang, Z.; Zhang, X.G.; Li, S.M.; Yang, Y.; Long, W.H. Genome-wide identification of the GS3 gene family and the influence of natural variations in BnGS3-3 on salt and cold stress tolerance in Brassica napus. Plants 2025, 14, 1145. [Google Scholar] [CrossRef]
- Wang, P.; Liu, F.; Wang, Y.F.; Chen, H.; Liu, T.; Li, M.Y.; Chen, S.J.; Wang, D.J. Deciphering crucial salt-responsive genes in Brassica napus via statistical modeling and network analysis on dynamic transcriptomic data. Plant Physiol. Biochem. 2025, 220, 109568. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, H.; Yang, Y.; Feng, X.; Chen, X.; Xiao, F.; Lin, H.; Guo, Y. Insights into plant salt stress signaling and tolerance. J. Genet. Genom. 2024, 51, 16–34. [Google Scholar] [CrossRef]
- Ashraf, M.; McNeilly, T. Salinity tolerance in Brassica oilseeds. Crit. Rev. Plant Sci. 2004, 23, 157–174. [Google Scholar] [CrossRef]
- Bhattarai, S.P.; Pendergast, L.; David, J. Root aeration improves yield and water use efficiency of tomato in heavy clay and saline soils. Sci. Hortic. 2006, 108, 278–288. [Google Scholar] [CrossRef]
- Hu, J.H.; Chen, B.Y.; Zhao, J.; Zhang, F.; Xie, T.; Xu, K.; Gao, G.; Yan, G.; Li, H.; Li, L.; et al. Genomic selection and genetic architecture of agronomic traits during modern rapeseed breeding. Nat. Genet. 2022, 54, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Zörb, C.; Geilfus, C.-M.; Dietz, K.-J. Salinity and crop yield. Plant Biol. 2019, 21, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Behera, L.M.; Hembram, P. Advances on plant salinity stress responses in the post-genomic era: A review. J. Crop Sci. Biotechnol. 2021, 24, 117–126. [Google Scholar] [CrossRef]
- Mohamed, I.A.A.; Shalby, N.; El-Badri, A.M.; Batool, M.; Wang, C.Y.; Wang, Z.K.; Salah, A.; Rady, M.M.; Jie, K.; Wang, B.; et al. RNA-seq analysis revealed key genes associated with salt tolerance in rapeseed germination through carbohydrate metabolism, hormone, and MAPK signaling pathways. Ind. Crops Prod. 2022, 176, 114262. [Google Scholar] [CrossRef]
- Guo, S.H.; Zhang, M.; Feng, M.X.; Liu, G.P.; Torregrosa, L.; Tao, X.Q.; Ren, R.H.; Fang, Y.L.; Zhang, Z.W.; Meng, J.F.; et al. miR156b-targeted VvSBP8/13 functions downstream of the abscisic acid signal to regulate anthocyanins biosynthesis in grapevine fruit under drought. Hortic. Res. 2024, 11, uhad293. [Google Scholar] [CrossRef]
- Gao, Z.; Nie, J.; Wang, H. MicroRNA biogenesis in plant. Plant Growth Regul. 2021, 93, 1–12. [Google Scholar] [CrossRef]
- Ai, B.; Chen, Y.; Zhao, M.; Ding, G.; Xie, J.; Zhang, F. Overexpression of miR1861h increases tolerance to salt stress in rice (Oryza sativa L.). Genet. Resour. Crop Evol. 2021, 68, 87–92. [Google Scholar] [CrossRef]
- Jung, H.J.; Kang, H. Expression and functional analyses of microRNA417 in Arabidopsis thaliana under stress conditions. Plant Physiol. Biochem. 2007, 45, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yang, L.; Zeng, H.Q.; Zhou, Z.S.; Yang, Z.M.; Li, H.; Sun, D.; Xie, F.; Zhang, B. A cotton miRNA is involved in regulation of plant response to salt stress. Sci. Rep. 2016, 6, 19736. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kwak, K.J.; Jung, H.J.; Lee, H.J.; Kang, H. MicroRNA402 affects seed germination of Arabidopsis thaliana under stress conditions via targeting DEMETER-LIKE protein3 mRNA. Plant Cell Physiol. 2010, 51, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhang, L.; Yang, Z.; Wei, Y.; Dong, T. Identification and functional characterization of plant miRNA under salt stress shed light on salinity resistance improvement through miRNA manipulation in crops. Front. Plant Sci. 2021, 12, 665439. [Google Scholar] [CrossRef]
- Zhan, J.; Diao, Y.; Yin, G.; Sajjad, M.; Wei, X.; Lu, Z.; Wang, Y. Integration of mRNA and miRNA analysis reveals the molecular mechanism of cotton response to salt stress. Front. Plant Sci. 2021, 12, 767984. [Google Scholar] [CrossRef]
- Kang, Y.; Yang, X.; Liu, Y.; Shi, M.; Zhang, W.; Fan, Y.; Yao, Y.; Zhang, J.; Qin, S. Integration of mRNA and miRNA analysis reveals the molecular mechanism of potato (Solanum tuberosum L.) response to alkali stress. Int. J. Biol. Macromol. 2021, 182, 938–949. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Chen, W.; Fu, Z.; Zhao, S.; E, Y.; Zhang, H.; Zhang, B.; Sun, M.; Han, P.; et al. Integration of mRNA and miRNA analysis reveals the molecular mechanisms of sugar beet (Beta vulgaris L.) response to salt stress. Sci. Rep. 2023, 13, 22074. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Q.; Liu, P.; Jia, W.; Chen, Z.; Xu, Z. Integration of mRNA and miRNA analysis reveals the molecular mechanism underlying salt and alkali stress tolerance in tobacco. Int. J. Mol. Sci. 2019, 20, 2391. [Google Scholar] [CrossRef]
- Zhou, B.; Gao, X.; Zhao, F. Integration of mRNA and miRNA analysis reveals the post-transcriptional regulation of salt stress response in Hemerocallis fulva. Int. J. Mol. Sci. 2023, 24, 7290. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-9 pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, J.; Xu, Q.; Dong, J.; Song, L.; Wang, W.; Shen, F. Long noncoding RNA lncRNA354 functions as a competing endogenous RNA of miR160b to regulate genes in response to salt stress in upland cotton. Plant Cell Environ. 2021, 44, 3302–3321. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.-J.; Tao, J.-J.; Cheng, T.; Bian, X.-H.; Wei, W.; Zhang, W.-K.; Ma, B.; Chen, S.-Y.; Zhang, J.-S. Soybean miR172a improves salt tolerance and can function as a long-distance signal. Mol. Plant 2016, 9, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, T.; Zhang, Y.; Li, Y. Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J. Exp. Bot. 2017, 68, 4727–4729. [Google Scholar] [PubMed]
- Sahito, Z.A.; Wang, L.; Sun, Z.; Yan, Q.; Zhang, X.; Jiang, Q.; Ullah, I.; Tong, Y.; Li, X. The miR172c-NNC1 module modulates root plastic development in response to salt in soybean. BMC Plant Biol. 2017, 17, 116. [Google Scholar] [CrossRef]
- Cheng, X.; He, Q.; Tang, S.; Wang, H.; Zhang, X.; Lv, M.; Liu, H.; Gao, Q.; Zhou, Y.; Wang, Q.; et al. The miR172/IDS1 signaling module confers salt tolerance through maintaining ROS homeostasis in cereal crops. New Phytol. 2021, 230, 1017–1033. [Google Scholar] [CrossRef]
- Song, J.B.; Gao, S.; Sun, D.; Li, H.; Shu, X.X.; Yang, Z.M. miR394 and LCR Are Involved in Arabidopsis Salt and Drought Stress Responses in an Abscisic Acid-Dependent Manner. BMC Plant Biol. 2013, 13, 210. [Google Scholar] [CrossRef]
- Hong, Y.C.; Guan, X.J.; Wang, X.; Kong, D.L.; Yu, S.J.; Wang, Z.Q.; Yu, Y.D.; Chao, Z.F.; Liu, X.; Huang, S.W.; et al. Natural variation in SlSOS2 promoter hinders salt resistance during tomato domestication. Hortic. Res. 2023, 10, uhac244. [Google Scholar] [CrossRef]
- Wan, H.; Qian, J.; Zhang, H.; Lu, H.; Li, O.; Li, R.; Yu, Y.; Wen, J.; Zhao, L.; Yi, B.; et al. Combined Transcriptomics and Metabolomics Analysis Reveals the Molecular Mechanism of Salt Tolerance of Huayouza 62, an Elite Cultivar in Rapeseed (Brassica napus L.). Int. J. Mol. Sci. 2022, 23, 1279. [Google Scholar] [CrossRef]
- Jin, Q.F.; Wang, Z.; Sandhu, D.; Chen, L.; Shao, C.Y.; Shang, F.H.; Xie, S.Y.; Huang, F.Y.; Chen, Z.Y.; Zhang, X.Q.; et al. mRNA-miRNA analyses reveal the involvement of CsbHLH1 and miR1446a in the regulation of caffeine biosynthesis in Camellia sinensis. Hortic. Res. 2024, 11, uhad282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yuan, S.; Zhou, M.; Yuan, N.; Li, Z.; Hu, Q.; Bethea, F.G., Jr.; Liu, H.; Li, S.; Luo, H. Transgenic creeping bent-grass overexpressing Osa-miR393a exhibits altered plant development and improved multiple stress tolerance. Plant Biotechnol. J. 2019, 17, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhou, Y.; Liu, X.; Meng, F.; Xu, C.; Chen, M. Integrated Transcriptomic and metabolomic analyses uncover the key pathways of Limonium bicolor in response to salt stress. Plant Biotechnol. J. 2025, 23, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Si, L.; Zhang, L.; Guo, R.; Wang, R.; Dong, H.; Guo, C. Metabolomics and transcriptomics analysis revealed the response mechanism of alfalfa to combined cold and saline-alkali stress. Plant J. 2024, 119, 1900–1919. [Google Scholar] [CrossRef]
- Chen, H.; Li, H.; Chong, X.; Zhou, T.; Lu, X.; Wang, X.; Zheng, B. Transcriptome Analysis of the Regulatory Mechanisms of Holly (Ilex dabieshanensis) under Salt Stress Conditions. Plants 2024, 13, 1638. [Google Scholar] [CrossRef]
- Salinas-Cornejo, J.; Madrid-Espinoza, J.; Verdugo, I.; Norambuena, L.; Ruiz-Lara, S. A SNARE-like Protein from Solanum lycopersicum Increases Salt Tolerance by Modulating Vesicular Trafficking in Tomato. Front. Plant Sci. 2023, 14, 1212806. [Google Scholar] [CrossRef]
- Farhangi-Abriz, S.; Ghassemi-Golezani, K. How Can Salicylic Acid and Jasmonic Acid Mitigate Salt Toxicity in Soybean Plants? Ecotoxicol. Environ. Saf. 2018, 147, 1010–1016. [Google Scholar] [CrossRef]
- Yang, R.; Yang, Z.; Xing, M.; Jing, Y.; Zhang, Y.; Zhang, K.; Zhou, Y.; Zhao, H.; Qiao, W.; Sun, J. TaBZR1 Enhances Wheat Salt Tolerance via Promoting ABA Biosynthesis and ROS Scavenging. J. Genet. Genom. 2023, 50, 861–871. [Google Scholar] [CrossRef]
- Cheng, Y.X.; Wang, Q.; Yang, L.X.; Li, Q.Z.; Yan, X.J. MiR319a-Mediated Salt Stress Response in Poplar. Hortic. Res. 2024, 11, uhae15. [Google Scholar] [CrossRef]
- Balasubramaniam, T.; Wijewardene, I.; Hu, R.; Shen, G.; Zhang, J.; Zhang, H. Co-Overexpression of AVP1, PP2A-C5, and AtCLCc in Arabidopsis Thaliana Greatly Increases Tolerance to Salt and Drought Stresses. Environ. Exp. Bot. 2022, 200, 104934. [Google Scholar] [CrossRef]
- Zhang, R.; Gong, R.; An, Z.; Li, G.; Dai, C.; Yi, R.; Liu, Y.; Dong, J.; Hu, J. Integrated physiological, transcriptomic and metabolomic analyses of glossy mutant under drought stress in rapeseed (Brassica napus L.). Ind. Crop Prod. 2025, 223, 120007. [Google Scholar] [CrossRef]
- Moore, S.; Stein, W.H. Chromatography of amino acids on sulfonated polystyrene resins. J. Biol. Chem. 1951, 192, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Mao, L.; Wei, X.; Xia, M.; Xu, C. MYB41, MYB107, and MYC2 promote ABA-mediated primary fatty alcohol accumulation via activation of AchnFAR in wound suberization in kiwifruit. Hortic. Res. 2020, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Arnon, D.I. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol. 1949, 24, 1–15. [Google Scholar] [CrossRef]
- Bakirov, A.; Zhang, Y.; Zhang, Q.; Seitahmetovna, S.A.; Yu, X.; Shi, Y.; Xu, Y.; Wang, K.; Qin, M.; Xu, A.; et al. Screening of Salt Tolerance Traits and the Salt Tolerance Evaluation Method in Brassica napus at the Seed Germination Stage. Ital. J. Agron. 2022, 17, 2011. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 Accurately Identifies Known and Hundreds of Novel microRNA Genes in Seven Animal Clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.; Chen, M. MTide: An Integrated Tool for the Identification of miRNA–Target Interaction in Plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq Gene Expression Estimation with Read Mapping Uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef]
- Dai, X.B.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 Release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New Features for Data Integration and Network Visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS Server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A Highly Sensitive RT-PCR Method for Detection and Quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Module of WGCNA | Gene Name | Gene ID | Category | GO ID | Term | Gene Ratio | p-Value |
|---|---|---|---|---|---|---|---|
| 24 h MEivory | TIFY6B | BnaC05G0424600ZS | Biological Process | GO: 2000022 | Regulation of the jasmonic acid-mediated signaling pathway | 25/1574 | 3.55 × 10−9 |
| 24 h MEivory | ABI5 | BnaA05G0087100ZS | Biological Process | GO: 0010029 | Regulation of seed germination | 29/1574 | 6.58 × 10−5 |
| 24 h MEivory | ABI5 | BnaA05G0087100ZS | Biological Process | GO: 1900140 | Regulation of seedling development | 29/1574 | 1.89 × 10−4 |
| 24 h MEivory | TPS9 | BnaA08G0235000ZS | Biological Process | GO: 0070413 | Trehalose metabolism in response to stress | 10/1574 | 2.45 × 10−4 |
| 24 h MEivory | RCD1 | BnaC05G0343300ZS | Biological Process | GO: 0072593 | Reactive oxygen species metabolic process | 28/1574 | 1.23 × 10−3 |
| 24 h MEivory | NHX7/ SOS1 | BnaC09G0255900ZS | Biological Process | GO: 0072593 | Reactive oxygen species metabolic process | 28/1574 | 1.23 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Li, D.; Qiao, Y.; Fan, N.; Gong, R.; Zhong, H.; Zhang, Y.; Lei, L.; Hu, J.; Dong, J. Integration of mRNA and miRNA Analysis Reveals the Regulation of Salt Stress Response in Rapeseed (Brassica napus L.). Plants 2025, 14, 2418. https://doi.org/10.3390/plants14152418
Liu Y, Li D, Qiao Y, Fan N, Gong R, Zhong H, Zhang Y, Lei L, Hu J, Dong J. Integration of mRNA and miRNA Analysis Reveals the Regulation of Salt Stress Response in Rapeseed (Brassica napus L.). Plants. 2025; 14(15):2418. https://doi.org/10.3390/plants14152418
Chicago/Turabian StyleLiu, Yaqian, Danni Li, Yutong Qiao, Niannian Fan, Ruolin Gong, Hua Zhong, Yunfei Zhang, Linfen Lei, Jihong Hu, and Jungang Dong. 2025. "Integration of mRNA and miRNA Analysis Reveals the Regulation of Salt Stress Response in Rapeseed (Brassica napus L.)" Plants 14, no. 15: 2418. https://doi.org/10.3390/plants14152418
APA StyleLiu, Y., Li, D., Qiao, Y., Fan, N., Gong, R., Zhong, H., Zhang, Y., Lei, L., Hu, J., & Dong, J. (2025). Integration of mRNA and miRNA Analysis Reveals the Regulation of Salt Stress Response in Rapeseed (Brassica napus L.). Plants, 14(15), 2418. https://doi.org/10.3390/plants14152418