
Development and Validation of Multi-Locus GWAS-Based KASP Markers for Maize Ustilago maydis Resistance

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
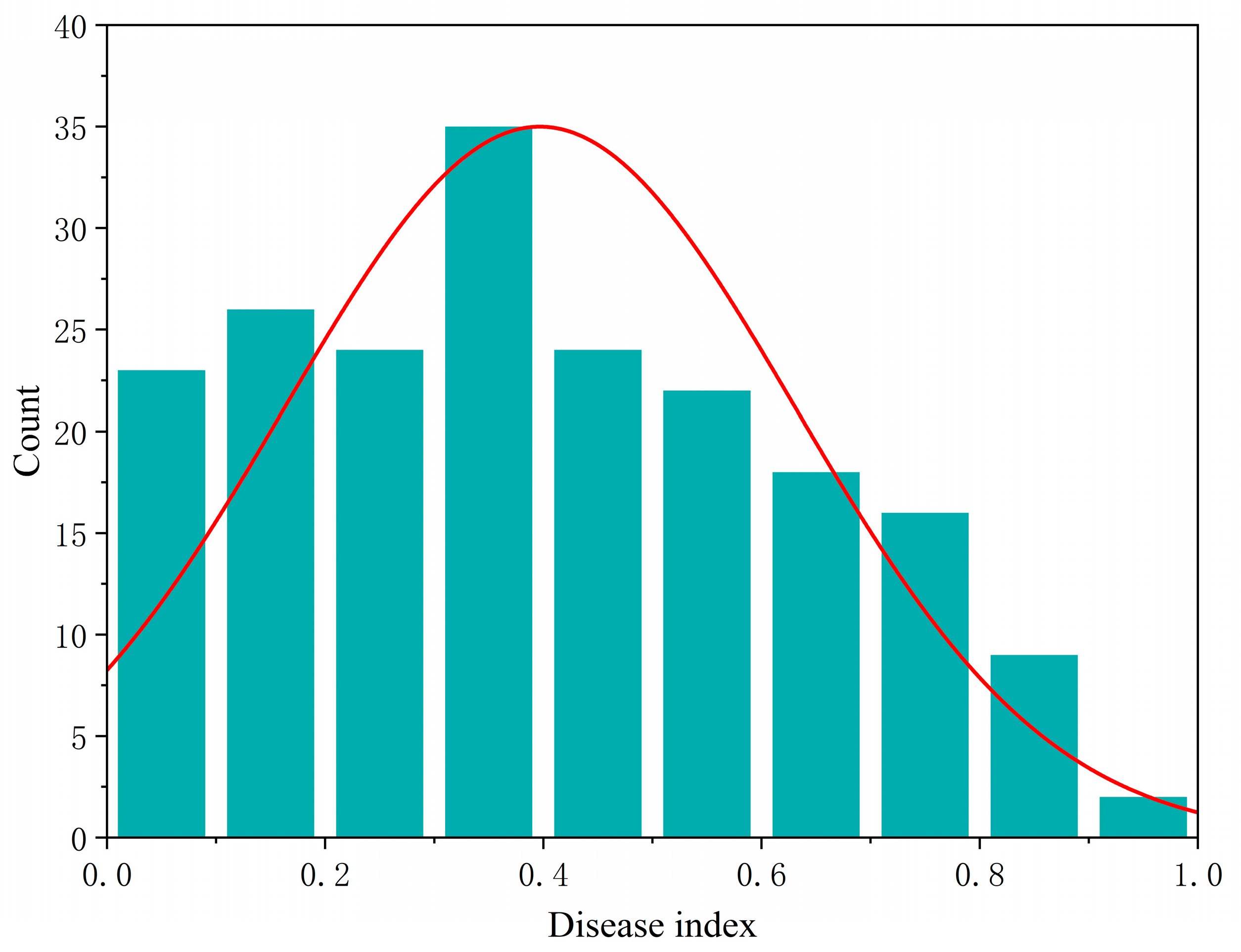
2.1. Analysis of Seedling U. maydis Resistance in Maize Inbred Lines
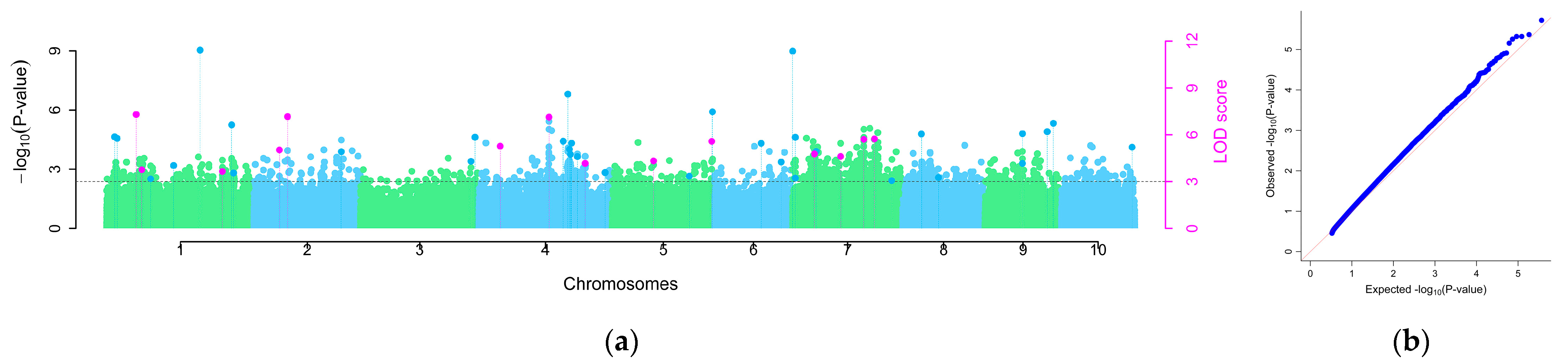
2.2. GWAS Analysis of Maize Inbred Lines’ Disease Indices
2.3. Identification of Candidate Genes for Maize U. maydis Resistance
2.4. Identification of Important SNP Loci and Candidate Genes
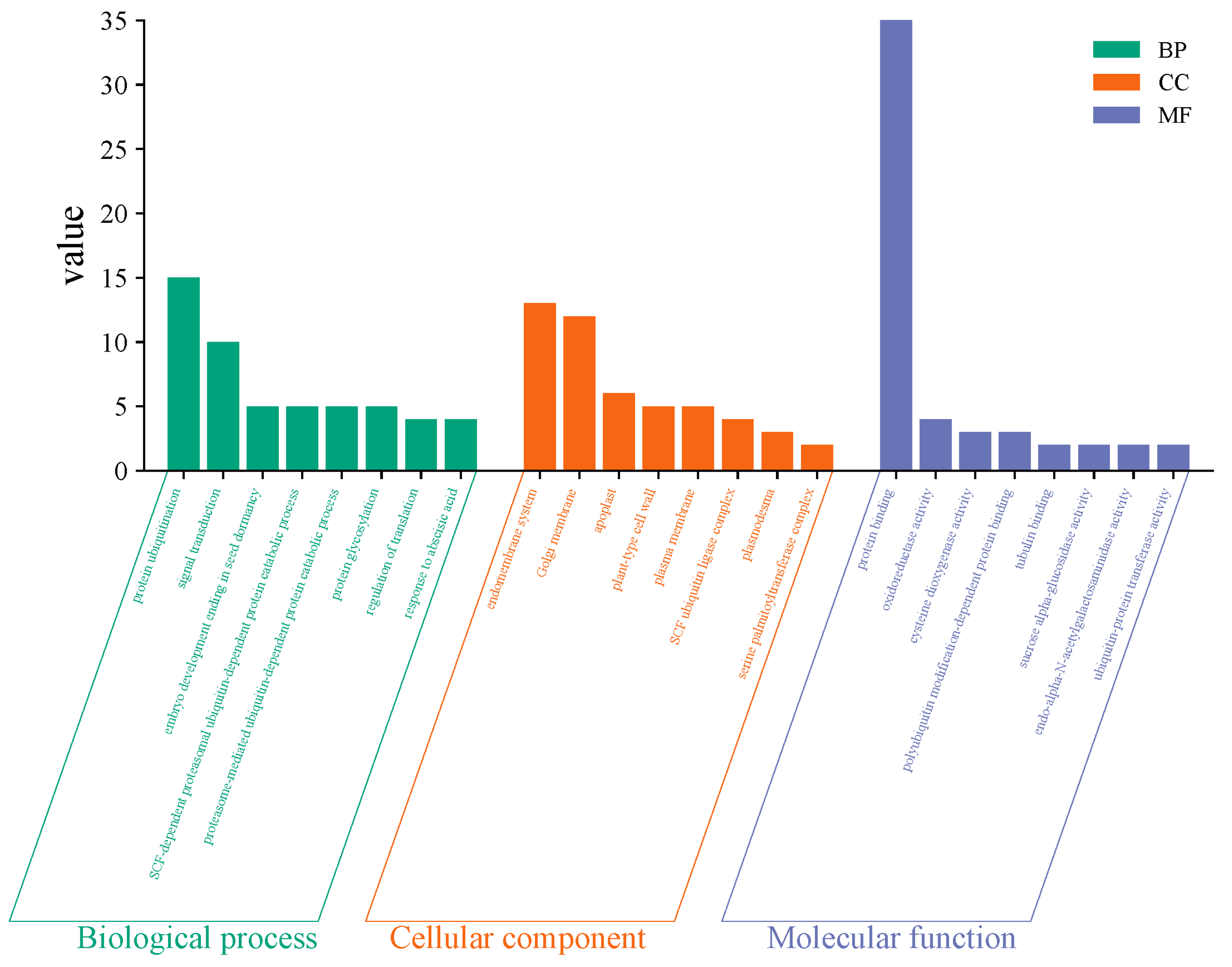
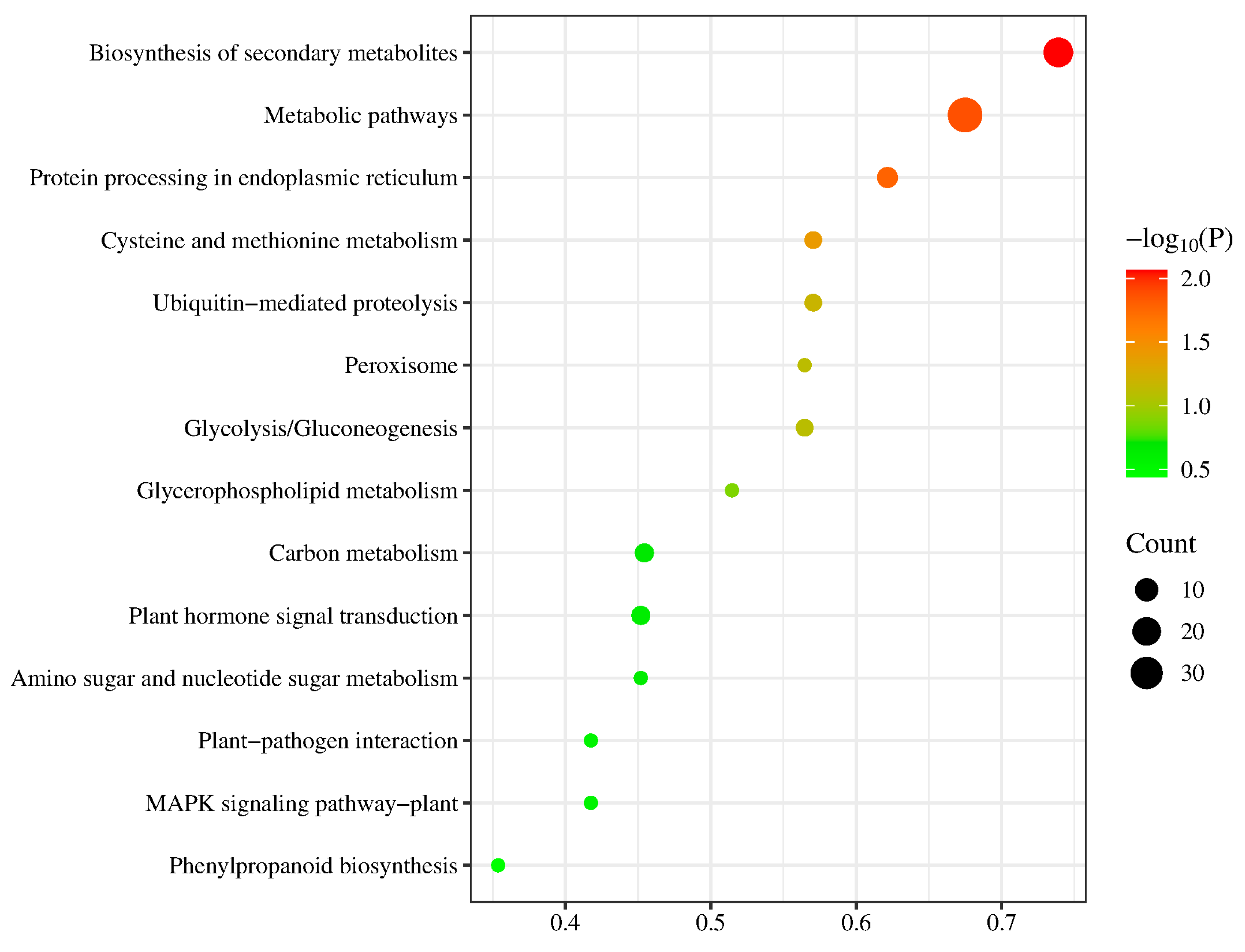
2.5. GO and KEGG
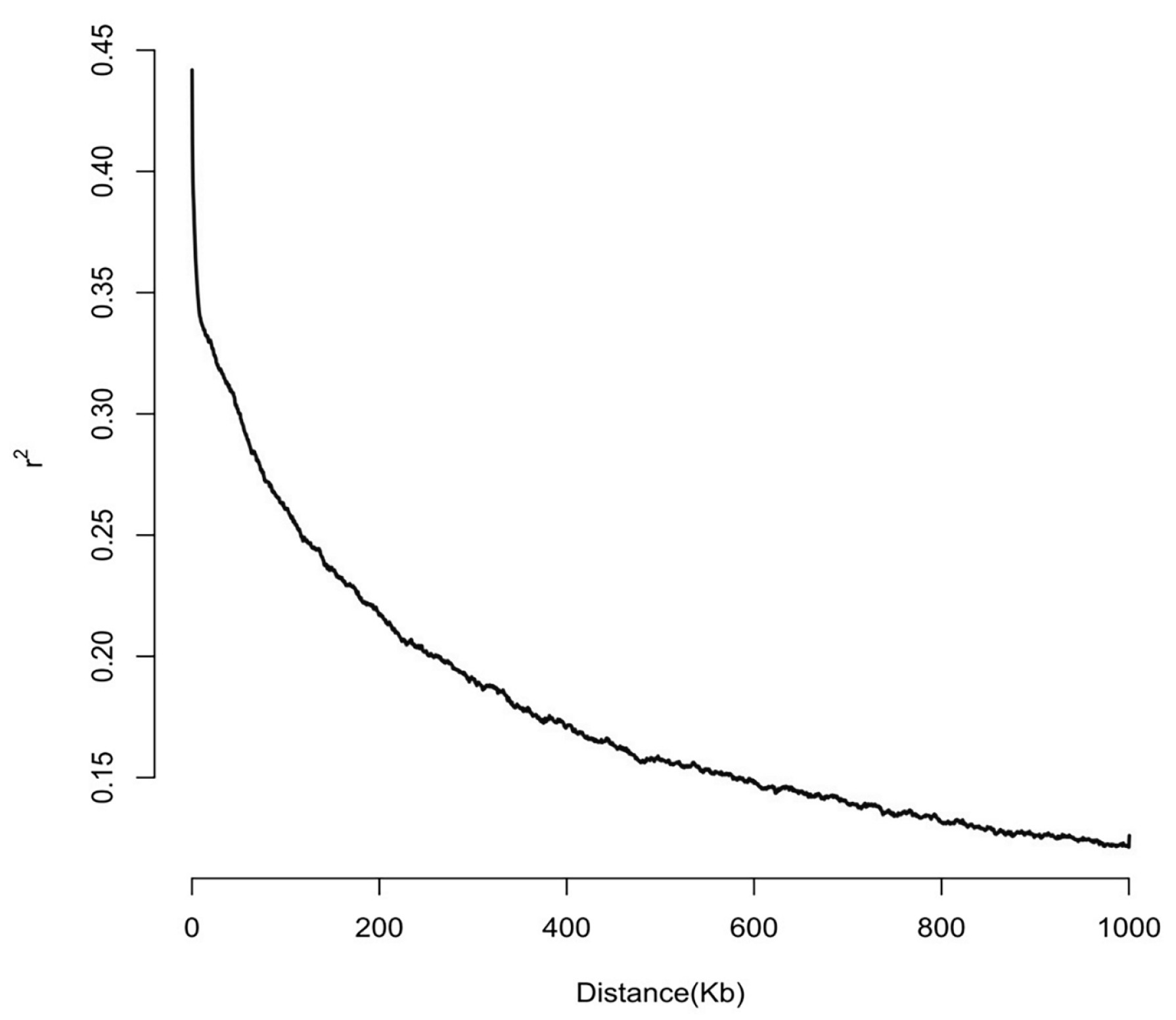
2.6. Haplotype Analysis
2.7. Design Development, Effect Analysis, and Validation of KASP Markers
2.7.1. Design and Development of the KASP Markers
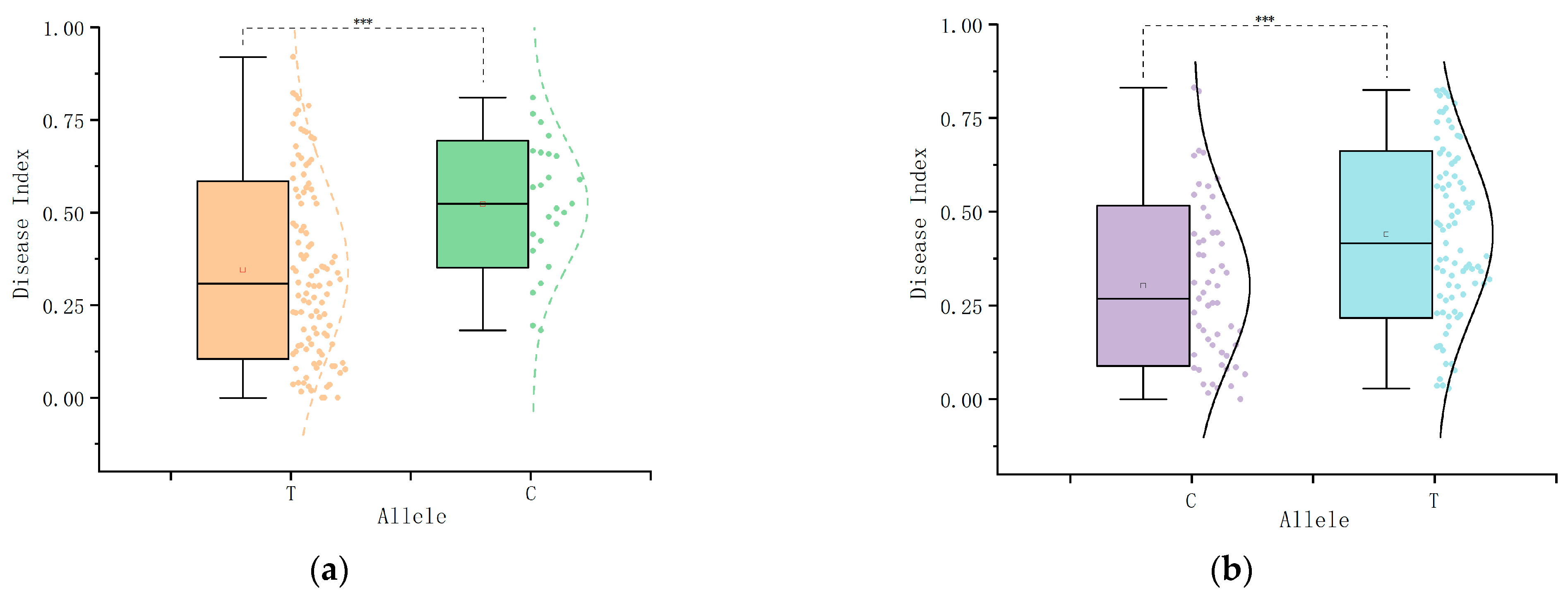
2.7.2. Utility Analysis of the KASP Markers
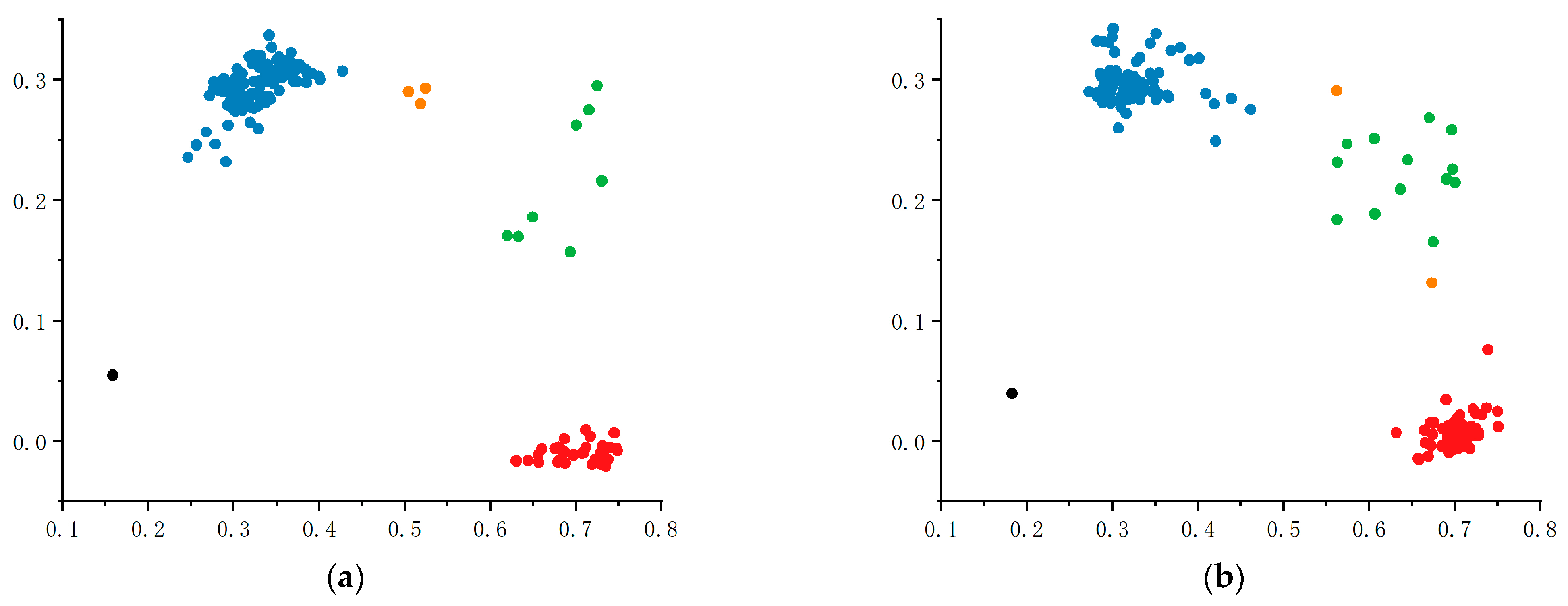
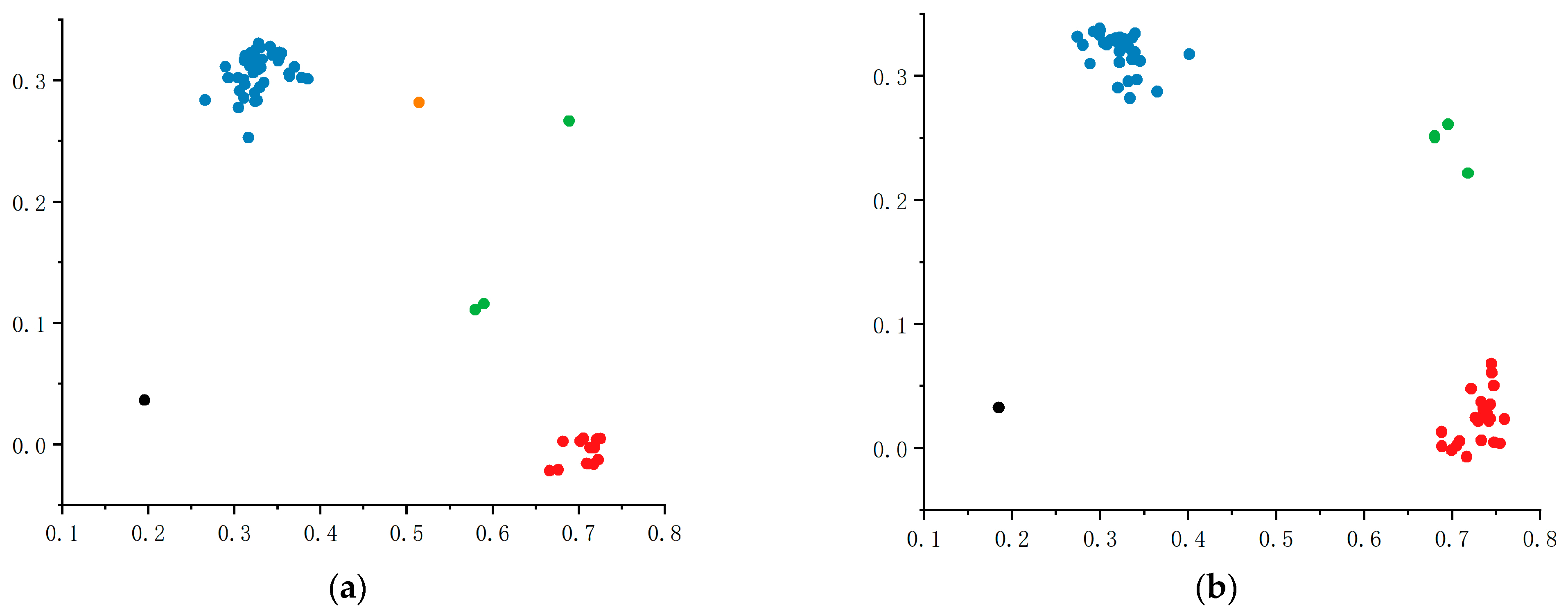
2.7.3. Validation of KASP Markers
2.7.4. Application of KASP Markers in the Selection of Highly Resistant Materials
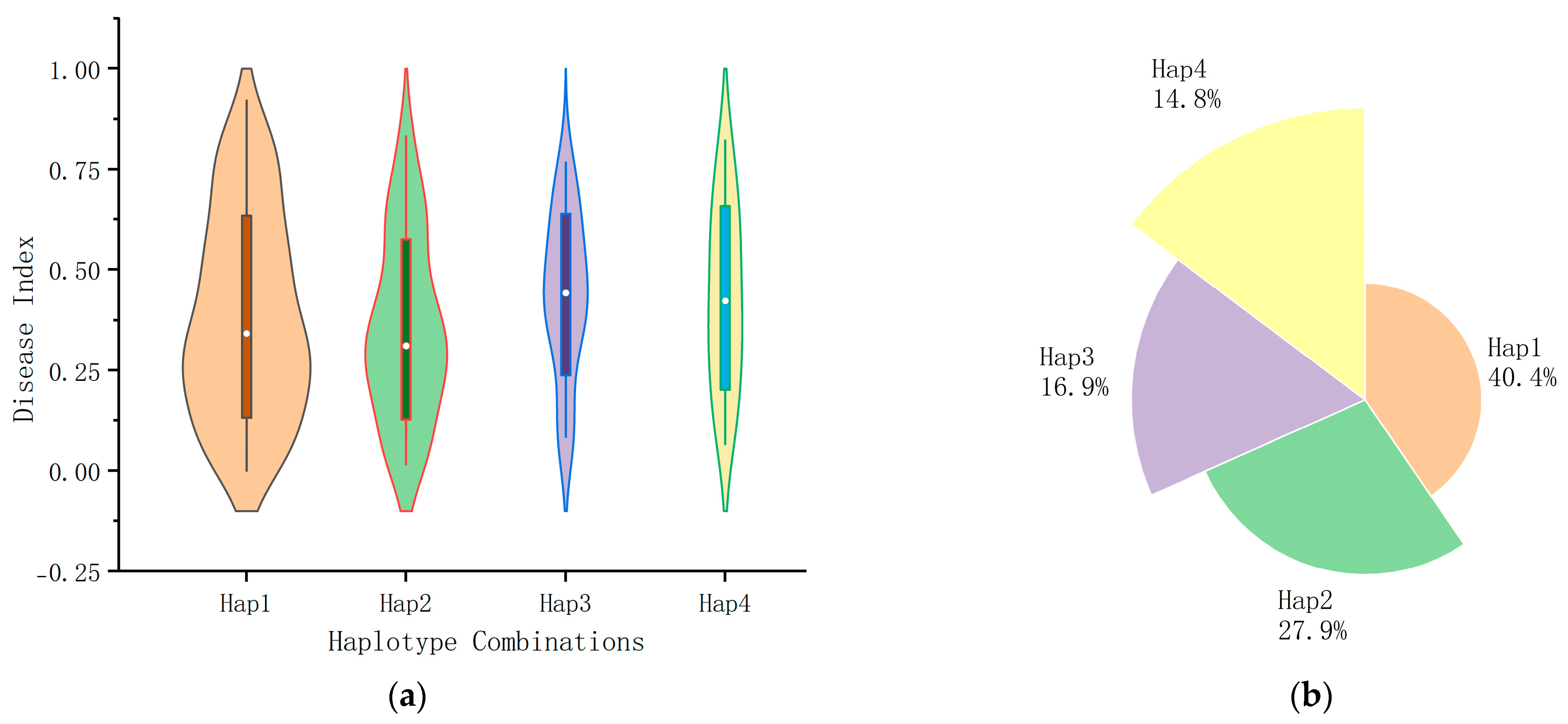
2.8. Effect of Haplotype Combinations Formed Based on KASP Markers for Maize U. maydis Resistance
3. Discussion
3.1. Maize U. maydis Resistance Identification and Resource Evaluation
3.2. GWAS-Significant Loci and Candidate Genes
3.3. Analysis of KASP Marker Applications
4. Materials and Methods
4.1. Experimental Materials
4.1.1. Experimental Maize Inbred Lines
4.1.2. Test Strains
4.2. Test Methods
4.2.1. Maize Cultivation and SG200 Culture
4.2.2. SG200 Inoculation, Phenotyping, and Evaluation Criteria
4.2.3. Extraction of Seedling DNA from Maize Inbred Lines
4.2.4. GWAS Analysis and Candidate Gene Mining
4.2.5. Haplotype Analysis
4.2.6. KASP Marker Primer Design and Genotyping
4.2.7. Statistical Analysis of Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, B.; Xu, L.; Li, J.; Zheng, Y.; Song, W.; Hou, P.; Zhu, L.; Jia, X.; Zha, Y.; Song, W.; et al. Integrated Transcriptome and GWAS Analysis to Identify Candidate Genes for Ustilago maydis Resistance in Maize. Agriculture 2024, 14, 958. [Google Scholar] [CrossRef]
- Ferris, A.C.; Walbot, V. Understanding Ustilago maydis infection of multiple maize organs. J. Fungi 2020, 7, 8. [Google Scholar] [CrossRef]
- Lanver, D.; Müller, A.N.; Happel, P.; Schweizer, G.; Haas, F.B.; Franitza, M.; Pellegrin, C.; Reissmann, S.; Altmüller, J.; Rensing, S.A.; et al. The biotrophic development of Ustilago maydis studied by RNA-seq analysis. Plant Cell 2018, 30, 300–323. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Ruan, X.; Ma, L.; Zhang, Y.; Wang, Q.; Gao, X. Dissection of the complex transcription and metabolism regulation networks associated with maize resistance to Ustilago maydis. Genes 2021, 12, 1789. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Ahn, H.-K.; Ding, P.; Jones, J.D.G. Mutual potentiation of plant immunity by cell-surface and intracellular receptors. Nature 2021, 592, 110–115. [Google Scholar] [CrossRef]
- Yu, C.; Qi, J.; Han, H.; Wang, P.; Liu, C. Progress in pathogenesis research of Ustilago maydis, and the metabolites involved along with their biosynthesis. Mol. Plant Pathol. 2023, 24, 495–509. [Google Scholar] [CrossRef]
- Lanver, D.; Tollot, M.; Schweizer, G.; Presti, L.L.; Reissmann, S.; Ma, L.S.; Schuster, M.; Tanaka, S.; Liang, L.; Ludwig, N.; et al. Ustilago maydis effectors and their impact on virulence. Nat. Rev. Microbiol. 2017, 15, 409–421. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.D.G.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 2004, 428, 764–767. [Google Scholar] [CrossRef]
- Melotto, M.; Underwood, W.; Koczan, J.; Nomura, K.; He, S.Y. Plant stomata function in innate immunity against bacterial invasion. Cell 2006, 126, 969–980. [Google Scholar] [CrossRef]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef]
- Tsuda, K.; Katagiri, F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol. 2010, 13, 459–465. [Google Scholar] [CrossRef]
- Thulasi Devendrakumar, K.; Li, X.; Zhang, Y. MAP kinase signalling: Interplays between plant PAMP-and effector-triggered immunity. Cell. Mol. Life Sci. 2018, 75, 2981–2989. [Google Scholar] [CrossRef]
- Schurack, S.; Depotter, J.R.; Gupta, D.; Thines, M.; Doehlemann, G. Comparative transcriptome profiling identifies maize line specificity of fungal effectors in the maize–Ustilago maydis interaction. Plant J. 2021, 106, 733–752. [Google Scholar] [CrossRef]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef]
- Rabe, F.; Ajami-Rashidi, Z.; Doehlemann, G.; Kahmann, R.; Djamei, A. Degradation of the plant defence hormone salicylic acid by the biotrophic fungus Ustilago maydis. Mol. Microbiol. 2013, 89, 179–188. [Google Scholar] [CrossRef]
- Redkar, A.; Matei, A.; Doehlemann, G. Insights into host cell modulation and induction of new cells by the corn smut Ustilago maydis. Front. Plant Sci. 2017, 8, 899. [Google Scholar] [CrossRef]
- Zou, K.; Li, Y.; Zhang, W.; Jia, Y.; Wang, Y.; Ma, Y.; Lv, X.; Xuan, Y.; Du, W. Early infection response of fungal biotroph Ustilago maydis in maize. Front. Plant Sci. 2022, 13, 970897. [Google Scholar] [CrossRef]
- Nisa, W.U.; Sandhu, S.; Nair, S.K.; Kaur, H.; Kumar, A.; Rashid, Z.; Saykhedkar, G.; Vikal, Y. Insights into maydis leaf blight resistance in maize: A comprehensive genome-wide association study in sub-tropics of India. BMC Genom. 2024, 25, 760. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Chang, F.; Yang, H.; Jiao, F.; Wang, F.; Peng, Y. Genetic analysis and identification of the candidate genes of maize resistance to Ustilago maydis by BSA-Seq and RNA-Seq. BMC Plant Biol. 2025, 25, 831. [Google Scholar] [CrossRef]
- Tomkowiak, A. Identification of SNP and SilicoDArT Markers and Characterization of Their Linked Candidate Genes Associated with Maize Smut Resistance. Int. J. Mol. Sci. 2024, 25, 11358. [Google Scholar] [CrossRef]
- Baumgarten, A.M.; Suresh, J.; May, G.; Phillips, R.L. Mapping QTLs contributing to Ustilago maydis resistance in specific plant tissues of maize. Theor. Appl. Genet. 2007, 114, 1229–1238. [Google Scholar] [CrossRef]
- Ganal, M.W.; Altmann, T.; Röder, M.S. SNP identification in crop plants. Curr. Opin. Plant Biol. 2009, 12, 211–217. [Google Scholar] [CrossRef]
- Ertiro, B.T.; Ogugo, V.; Worku, M.; Das, B.; Olsen, M.; Labuschagne, M.; Semagn, K. Comparison of Kompetitive Allele Specific PCR (KASP) and genotyping by sequencing (GBS) for quality control analysis in maize. BMC Genom. 2015, 16, 908. [Google Scholar] [CrossRef]
- Jagtap, A.B.; Vikal, Y.; Johal, G.S. Genome-wide development and validation of cost-effective KASP marker assays for genetic dissection of heat stress tolerance in maize. Int. J. Mol. Sci. 2020, 21, 7386. [Google Scholar] [CrossRef]
- Cheon, K.S.; Jeong, Y.M.; Oh, H.; Oh, J.; Kang, D.Y.; Kim, N.; Lee, E.; Baek, J.; Kim, S.L.; Choi, I.; et al. Development of 454 new Kompetitive Allele-Specific PCR (KASP) markers for temperate japonica rice varieties. Plants 2020, 9, 1531. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Q.; Kang, Z.; Liu, S.; Li, H.; Mu, J.; Dai, M.; Han, D.; Zeng, Q.; Chen, X.; et al. Development and validation of KASP-SNP markers for QTL underlying resistance to stripe rust in common wheat cultivar P10057. Plant Dis. 2017, 101, 2079–2087. [Google Scholar] [CrossRef]
- Tan, C.; Assanga, S.; Zhang, G.; Rudd, J.C.; Haley, S.D.; Xue, Q.; Ibrahim, A.; Bai, G.; Zhang, X.; Byrne, P.; et al. Development and validation of KASP markers for wheat streak mosaic virus resistance gene Wsm2. Crop Sci. 2017, 57, 340–349. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, M.; Zhang, H.; Liu, X.; Zhang, W.; Wang, Q.; Jia, Q.; Xu, D.; Chen, H.; Su, C. A genome-wide association analysis for salt tolerance during the soybean germination stage and development of KASP markers. Front. Plant Sci. 2024, 15, 1352465. [Google Scholar] [CrossRef]
- Jia, Q.; Zhou, M.; Xiong, Y.; Wang, J.; Xu, D.; Zhang, H.; Liu, X.; Zhang, W.; Wang, Q.; Sun, X.; et al. Development of KASP markers assisted with soybean drought tolerance in the germination stage based on GWAS. Front. Plant Sci. 2024, 15, 1352379. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, W.; Cui, Y.; Sang, X.; Lu, J.; Jing, H.; Wang, W.; Zhao, P.; Wang, H. Detection of candidate genes and development of KASP markers for Verticillium wilt resistance by combining genome-wide association study, QTL-seq and transcriptome sequencing in cotton. Theor. Appl. Genet. 2021, 134, 1063–1081. [Google Scholar] [CrossRef]
- Wang, T.; Wang, T.; Wang, P.; Feng, Z.; Jin, Z.; Wang, F.; Wang, W.; Peng, Y. Resistance Identification and Genetic Diversity Analysis of Maize Germplasm Resources to Ustilago maydis. J. Maize Sci. 2020, 30, 30–38. [Google Scholar] [CrossRef]
- Zheng, Y.; Hou, P.; Zhu, L.; Song, W.; Liu, H.; Huang, Y.; Wang, H.; Guo, J. Genome-wide association study of vascular bundle-related traits in maize stalk. Front. Plant Sci. 2021, 12, 699486. [Google Scholar] [CrossRef]
- Zill, P.; Büttner, A.; Eisenmenger, W.; Möller, H.-J.; Bondy, B.; Ackenheil, M. Single nucleotide polymorphism and haplotype analysis of a novel tryptophan hydroxylase isoform (TPH2) gene in suicide victims. Biol. Psychiatry 2004, 56, 581–586. [Google Scholar] [CrossRef]
- Yan, L.; Li, Z.; Chen, J.; Gao, C.; Yu, Y.; Yan, Z. Primary identification of resistance in different maize varieties to Ustilago maydis. J. Hunan Agric. Univ. 2017, 43, 42–46. [Google Scholar] [CrossRef]
- Lv, X.; Li, X.C.; Hao, Z.; Ji, H.; Shi, L.; Zhang, S. Comparative QTL mapping of resistance to sugarcane mosaic virus in maize based on bioinformatics. Yi Chuan Hered. 2008, 30, 101–108. [Google Scholar] [CrossRef]
- Qiu, H.; Tan, K.; Li, C.; Shen, T.; Zhang, Z. Identification of QTL for resistance to head smut in maize (Zea mays. L). Euphytica 2021, 217, 185. [Google Scholar] [CrossRef]
- Pè, M.E.; Gianfranceschi, L.; Taramino, G.; Tarchini, R.; Angelini, P.; Dani, M.; Binelli, G. Mapping quantitative trait loci (QTLs) for resistance to Gibberella zeae infection in maize. Mol. Genet. Genom. 1993, 241–241, 11–16. [Google Scholar] [CrossRef]
- Robertson-Hoyt, L.A.; Jines, M.P.; Balint-Kurti, P.J.; Kleinschmidt, C.E.; White, D.G.; Payne, G.A.; Maragos, C.M.; Molnár, T.L.; Holland, J.B. QTL mapping for Fusarium ear rot and fumonisin contamination resistance in two maize populations. Crop Sci. 2006, 46, 1734–1743. [Google Scholar] [CrossRef]
- Berger, D.K.; Carstens, M.; Korsman, J.N.; Middleton, F.; Kloppers, F.J.; Tongoona, P.; Myburg, A.A. Mapping QTL conferring resistance in maize to gray leaf spot disease caused by Cercospora zeina. BMC Genet. 2014, 15, 60. [Google Scholar] [CrossRef]
- Dietrich, C.R.; Perera, M.A.D.N.; Yandeau-Nelson, M.D.; Meeley, R.B.; Nikolau, B.J.; Schnable, P.S. Characterization of two GL8 paralogs reveals that the 3-ketoacyl reductase component of fatty acid elongase is essential for maize (Zea mays L.) development. Plant J. 2005, 42, 844–861. [Google Scholar] [CrossRef]
- Bartholomew, E.S.; Black, K.; Feng, Z.; Liu, W.; Shan, N.; Zhang, X.; Wu, L.; Bailey, L.; Zhu, N.; Qi, C.; et al. Comprehensive analysis of the chitinase gene family in cucumber (Cucumis sativus L.): From gene identification and evolution to expression in response to Fusarium oxysporum. Int. J. Mol. Sci. 2019, 20, 5309. [Google Scholar] [CrossRef]
- Cheng, H.; Shao, Z.; Lu, C.; Duan, D. Genome-wide identification of chitinase genes in Thalassiosira pseudonana and analysis of their expression under abiotic stresses. BMC Plant Biol. 2021, 21, 87. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, X.; Li, L.; He, Y.; Hong, G.; Li, J.; Lin, L.; Cheng, Y.; Yan, F.; Chen, J.; et al. Suppression of auxin signalling promotes rice susceptibility to Rice black streaked dwarf virus infection. Mol. Plant Pathol. 2019, 20, 1093–1104. [Google Scholar] [CrossRef]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef]
- Liu, P.; Koizuka, N.; Martin, R.C.; Nonogaki, H. The BME3 (Blue Micropylar End 3) GATA zinc finger transcription factor is a positive regulator of Arabidopsis seed germination. Plant J. 2005, 44, 960–971. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, T.; Yin, K.-Q.; Chen, Z.; Gu, H.; Qu, L.J.; Qin, J. Arabidopsis RAP2. 2 plays an important role in plant resistance to Botrytis cinerea and ethylene responses. New Phytol. 2012, 195, 450–460. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Zhang, A.; Kong, D.; Liu, G.; Luo, L.; Yu, X. Development and validation of functional markers (tetra-primer ARMS and KASP) for the bacterial blight resistance gene xa5 in rice. Australas. Plant Pathol. 2021, 50, 323–327. [Google Scholar] [CrossRef]
- Fang, T.; Lei, L.; Li, G.; Powers, C.; Hunger, R.M.; Carver, B.F.; Yan, L. Development and deployment of KASP markers for multiple alleles of Lr34 in wheat. Theor. Appl. Genet. 2020, 133, 2183–2195. [Google Scholar] [CrossRef]
- Zhou, G.; Ma, L.; Zhao, C.; Xie, F.; Xu, Y.; Wang, Q.; Hao, D.; Gao, X. Genome-wide association study and molecular marker de-velopment for susceptibility to Gibberella ear rot in maize. Theor. Appl. Genet. 2024, 137, 222. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.-J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Morrison, E.N.; Emery, R.J.N.; Saville, B.J. Fungal derived cytokinins are necessary for normal Ustilago maydis infection of maize. Plant Pathol. 2017, 66, 726–742. [Google Scholar] [CrossRef]
- Brefort, T.; Doehlemann, G.; Mendoza-Mendoza, A.; Reissmann, S.; Djamei, A.; Kahmann, R. Ustilago maydis as a pathogen. Annu. Rev. Phytopathol. 2009, 47, 423–445. [Google Scholar] [CrossRef]
- Steinberg, G.; Perez-Martin, J. Ustilago maydis, a new fungal model system for cell biology. Trends Cell Biol. 2008, 18, 61–67. [Google Scholar] [CrossRef]
- Pathi, K.M.; Rink, P.; Budhagatapalli, N.; Betz, R.; Saado, I.; Hiekel, S.; Becker, M.; Djamei, A.; Kumlehn, J. Engineering smut resistance in maize by site-directed mutagenesis of LIPOXYGENASE 3. Front. Plant Sci. 2020, 11, 543895. [Google Scholar] [CrossRef]
- Zhang, J.; Stewart, J.M. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Yi, N.; Xu, S. Bayesian LASSO for quantitative trait loci mapping. Genetics 2008, 179, 1045–1055. [Google Scholar] [CrossRef]
- Wu, T.T.; Chen, Y.F.; Hastie, T.; Sobel, E.; Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009, 25, 714–721. [Google Scholar] [CrossRef]
- Ayers, K.L.; Cordell, H.J. SNP selection in genome-wide and candidate gene studies via penalized logistic regression. Genet. Epidemiol. 2010, 34, 879–891. [Google Scholar] [CrossRef]
- Wang, D.; Eskridge, K.M.; Crossa, J. Identifying QTLs and epistasis in structured plant populations using adaptive mixed LASSO. J. Agric. Biol. Environ. Stat. 2010, 16, 170–184. [Google Scholar] [CrossRef]
- Giglio, C.; Brown, S.D. Using elastic net regression to perform spectrally relevant variable selection. J. Chemom. 2018, 32, e3034. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4. 0.2: An R platform for multi-locus genome-wide association studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, F.; Zhou, Y. The application of multi-locus GWAS for the detection of salt-tolerance loci in rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.-Y.; Ni, Y.-L.; Wen, Y.-J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.-M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef]
- Ren, W.-L.; Wen, Y.-J.; Dunwell, J.M.; Zhang, Y.-M. pKWmEB: Integration of Kruskal–Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Wen, Y.-J.; Zhang, Y.-W.; Zhang, J.; Feng, J.-Y.; Dunwell, J.M.; Zhang, Y.-M. An efficient multi-locus mixed model framework for the detection of small and linked QTLs in F2. Brief. Bioinform. 2019, 20, 1913–1924. [Google Scholar] [CrossRef]
- Tamba, C.L.; Zhang, Y. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018. biorxiv:341784. [Google Scholar] [CrossRef]
- Sahito, J.H.; Zhang, H.; Gishkori, Z.G.N.; Ma, C.; Wang, Z.; Ding, D.; Zhang, X.; Tang, J. Advancements and prospects of genome-wide association studies (GWAS) in maize. Int. J. Mol. Sci. 2024, 25, 1918. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, T.; Qiao, W.; Liu, W.; Qiao, Y.; Li, Y.; Liu, M.; Ma, Y.; Dong, B. Optimizing canopy spacing configuration enhances foxtail millet grain yield and water productivity by improving stalk lodging resistance in the North China Plain. Eur. J. Agron. 2024, 158, 127230. [Google Scholar] [CrossRef]
- Bratianu, C.; Stanescu, D.F.; Mocanu, R. Exploring the knowledge management impact on business education. Sustainability 2021, 13, 2313. [Google Scholar] [CrossRef]
- Zheng, Y.; Yuan, F.; Huang, Y.; Zhao, Y.; Jia, X.; Zhu, L.; Guo, J. Genome-wide association studies of grain quality traits in maize. Sci. Rep. 2021, 11, 9797. [Google Scholar] [CrossRef]
- Mulugeta, B.; Tesfaye, K.; Keneni, G.; Ahmed, S. Genetic diversity in spring faba bean (Vicia faba L.) genotypes as revealed by high-throughput KASP SNP markers. Genet. Resour. Crop. Evol. 2021, 68, 1971–1986. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, R.; Pi, X.; Zhang, B. Structural features of rice starch-protein system: Influence of retrogradation time and quick-freezing temperature. Int. J. Biol. Macromol. 2024, 277, 133981. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Max | Min | Mean | SD | Variance | Skewness | Kurtosis | CV% | G × E | H2% |
|---|---|---|---|---|---|---|---|---|---|
| 0.9207 | 0.0167 | 0.3991 | 0.2294 | 0.0530 | 0.1870 | −0.9550 | 57.46 | 0.0016 | 74.94 |
| Significant SNP Locus | LOD Score | Model | Genome | Functional Notes |
|---|---|---|---|---|
| 1_244281660 | 8.57 6.43 | FASTmrMLM pKWmEB | Zm00001d032946 | Chitinase 1 |
| Zm00001d032948 | fatty acid elongase | |||
| 2_87571650 | 4.70 3.42 3.85 14.44 | FASTmrMLM FASTmrEMMA pLARmEB pKWmEB ISIS E-BLASSO | Zm00001d004159 | SNARE-interacting protein KEULE |
| 5_220156746 | 5.95 3.02 6.87 3.43 | FASTmrMLM FASTmrEMMA pLARmEB pKWmEB | Zm00001d018414 | IAA9—auxin-responsive Aux/IAA family member |
| Zm00001d018421 | GATA transcription factor 8 | |||
| 7_88700440 | 11.48 4.50 13.11 4.60 | FASTmrMLM FASTmrEMMA pKWmEB ISIS EM-BLASSO | Zm00001d020043 | AP2-EREBP transcription factor |
| Marker Name | Primer Name | Primer Sequence, 5′-3′ |
|---|---|---|
| qSB1-KASP | 1-A | GAAGGTGACC AAGTTCATGC TGACATCTGT CTACGACTAG AGCG |
| 1-B | GAAGGTCGGA GTCAACGGAT TGACATCTGT CTACGACTAG AGCA | |
| 1-C | TGATCGAACG ATGAGCCCGA TCATA | |
| qSB5-KASP | 5-A | GAAGGTGACC AAGTTCATGC TCAGCCCATA CCAAGTGGCT AG |
| 5-B | GAAGGTCGGA GTCAACGGAT TGCAGCCCAT ACCAAGTGGC TAA | |
| 5-C | GAGGTGAGAG ATACCACAGT CCTAT |
| Marker Name | Alleles (Number of Samples) | Mean Value of Disease Index | Selection Rate (Sample Size) | p-Value |
|---|---|---|---|---|
| qSB1-KASP | T (133) | 0.3720 | 48.12% (64/133) | p < 0.05 |
| C (45) | 0.4272 | 26.66% (12/45) | ||
| qSB5-KASP | T (92) | 0.4021 | 33.70% (31/92) | p < 0.05 |
| C (90) | 0.3776 | 43.33% (39/90) |
| SNP Significant Loci | Percentages of Favorable Alleles | DM07 | 18 | W668 | Lo1125 | Jingnuo2 | DH65232 |
|---|---|---|---|---|---|---|---|
| 1_244281660 (T/C) | 94.87% (37/39) | T | T | T | T | T | T |
| 5_220156746 (C/T) | 53.84% (21/39) | C | C | C | C | C | C |
| Marker Name | Hap1 | Hap2 | Hap3 | Hap4 |
|---|---|---|---|---|
| qSB1-KASP | T | T | C | C |
| qSB5-KASP | T | C | T | C |
| Haplotype Combinations (Sample Size) | Average Disease Index | Selection Rate (Sample Size) | p-Value |
|---|---|---|---|
| Hap1 (74) | 0.3828 | 43.24% (32) | p < 0.05 |
| Hap2 (51) | 0.3423 | 58.82% (30) | |
| Hap3 (32) | 0.4380 | 16.67% (3) | |
| Hap4 (27) | 0.4302 | 25.92% (7) |
| Level of Disease | Performance Symptoms |
|---|---|
| 1 | No infection symptoms are observed in the plants |
| 2 | Minimal infection manifests as chromatic alterations on inoculated leaves |
| 3 | Moderate infection produces pathognomonic rice-grain-sized tumors on foliar and sheath tissues |
| 4 | Severe infections cause pathological distortion of leaves, stems, and basal culms, accompanied by enlarged verrucous proliferations |
| Disease Index | Capability to Resist | Abbreviated |
|---|---|---|
| 0–15.0 | Highly resistant | HR |
| 15.1–30.0 | Resistant | R |
| 30.1–50.0 | Moderately resistant | MR |
| 50.1–70.0 | Susceptible | S |
| 70.1–100.0 | Highly susceptible | HS |
| PCR Reaction Components | System Usage (µL) |
|---|---|
| DNA | 1.5 |
| 2× Master mix | 0.75 |
| KASP marker primer | 0.0417 |
| H2O | 0.75 |
| Total volume | 3 |
| Steps | Effect | Temperature (°C) | Time | Number of Cycles |
|---|---|---|---|---|
| 1 | Denaturation | 94 | 15 min | 1 |
| 2 | Denaturation | 94 | 20S | 10 |
| Annealing/Extension | 61–55 | 60S | ||
| 3 | Denaturation | 94 | 20S | 26 |
| Annealing/Extension | 55 | 60S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, T.; Gao, H.; Wang, C.; Zheng, Y.; Song, W.; Hou, P.; Zhu, L.; Zhao, Y.; Song, W.; Guo, J. Development and Validation of Multi-Locus GWAS-Based KASP Markers for Maize Ustilago maydis Resistance. Plants 2025, 14, 2315. https://doi.org/10.3390/plants14152315
Shen T, Gao H, Wang C, Zheng Y, Song W, Hou P, Zhu L, Zhao Y, Song W, Guo J. Development and Validation of Multi-Locus GWAS-Based KASP Markers for Maize Ustilago maydis Resistance. Plants. 2025; 14(15):2315. https://doi.org/10.3390/plants14152315
Chicago/Turabian StyleShen, Tao, Huawei Gao, Chao Wang, Yunxiao Zheng, Weibin Song, Peng Hou, Liying Zhu, Yongfeng Zhao, Wei Song, and Jinjie Guo. 2025. "Development and Validation of Multi-Locus GWAS-Based KASP Markers for Maize Ustilago maydis Resistance" Plants 14, no. 15: 2315. https://doi.org/10.3390/plants14152315
APA StyleShen, T., Gao, H., Wang, C., Zheng, Y., Song, W., Hou, P., Zhu, L., Zhao, Y., Song, W., & Guo, J. (2025). Development and Validation of Multi-Locus GWAS-Based KASP Markers for Maize Ustilago maydis Resistance. Plants, 14(15), 2315. https://doi.org/10.3390/plants14152315