

Genome-Wide Analysis of GmMYB S20 Transcription Factors Reveals Their Critical Role in Soybean Nodulation
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
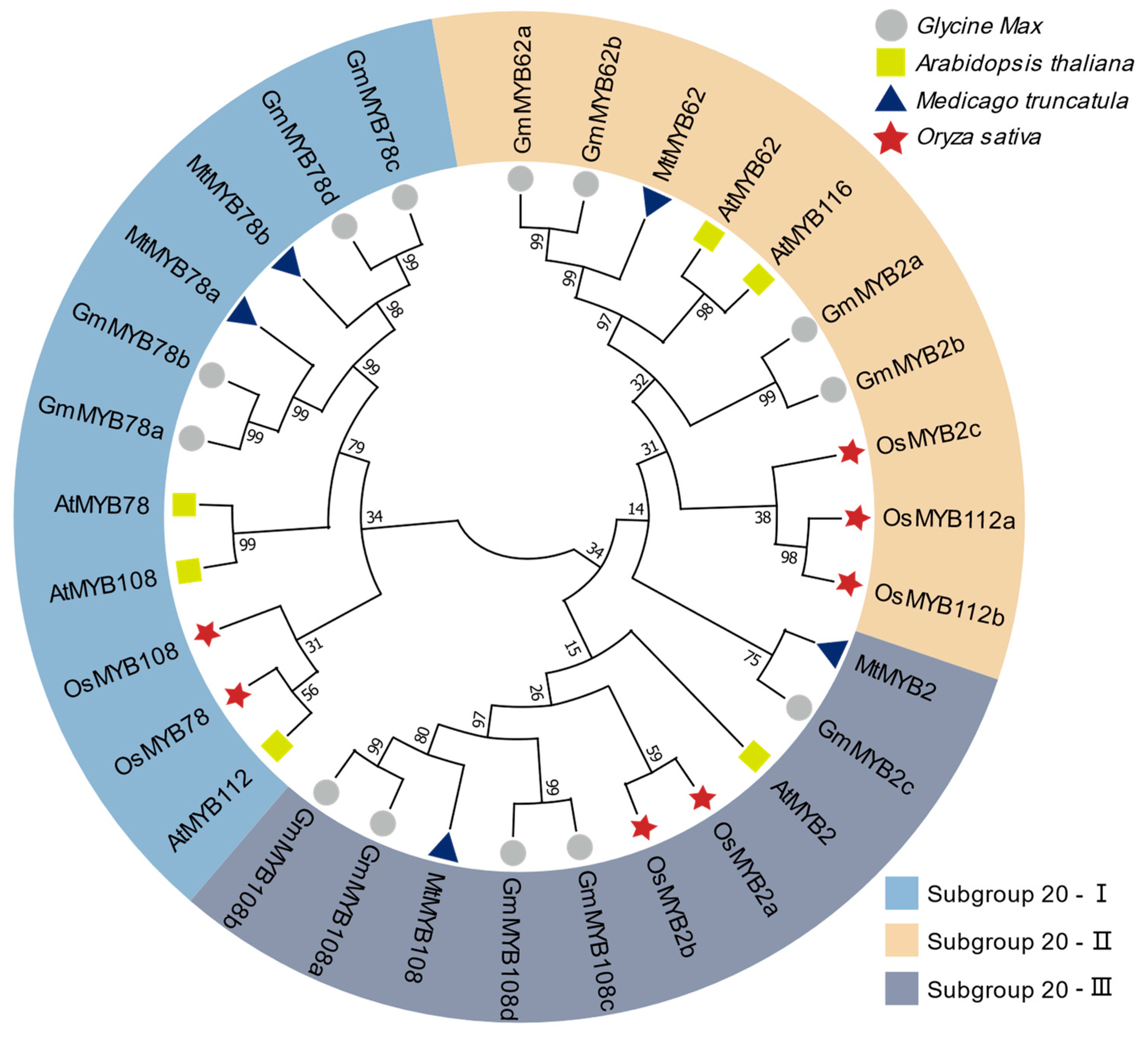
2.1. Classification and Phylogenetic Analysis of MYB Subgroup 20 Genes
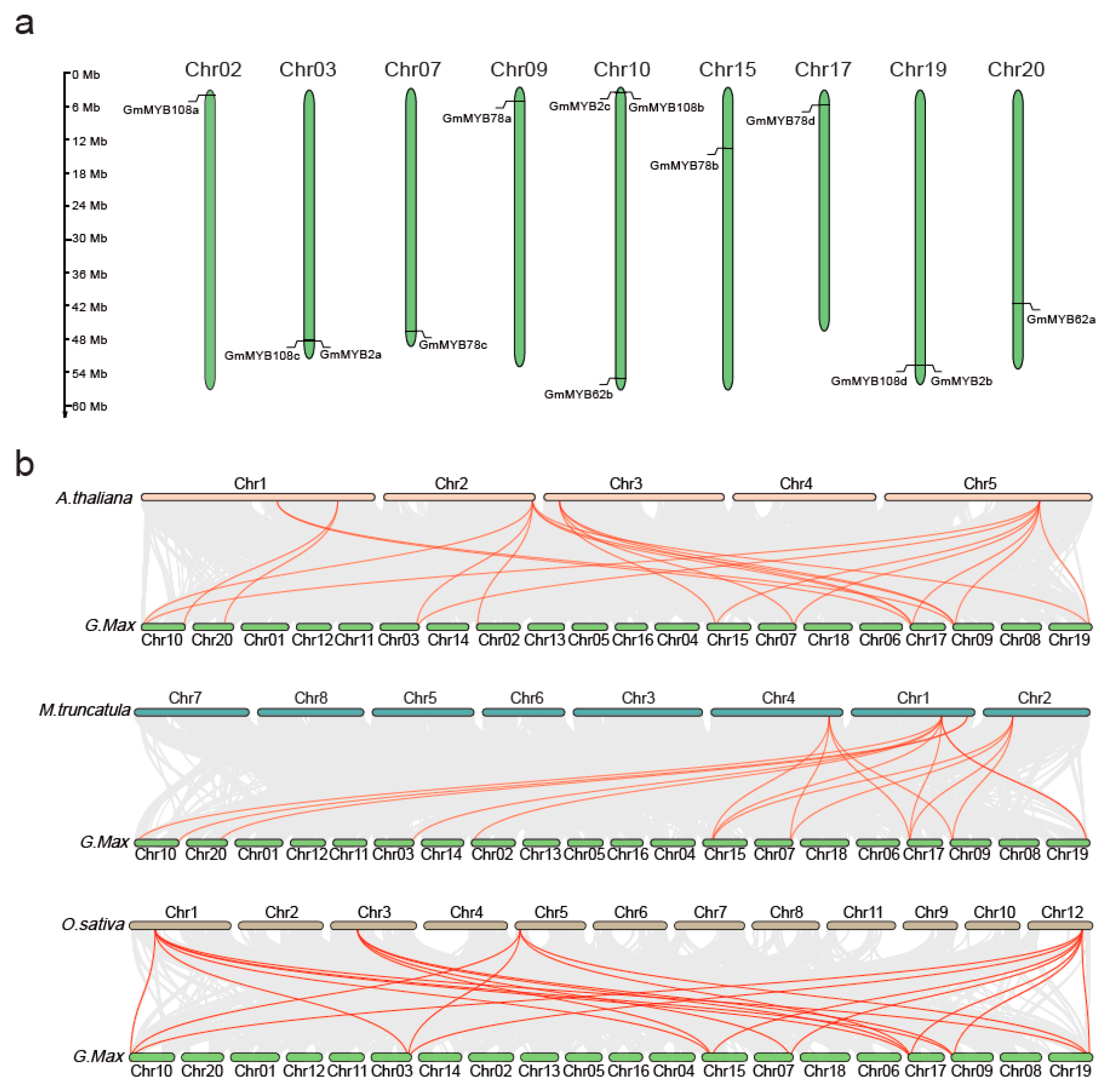
2.2. Chromosomal Mapping of GmMYB S20
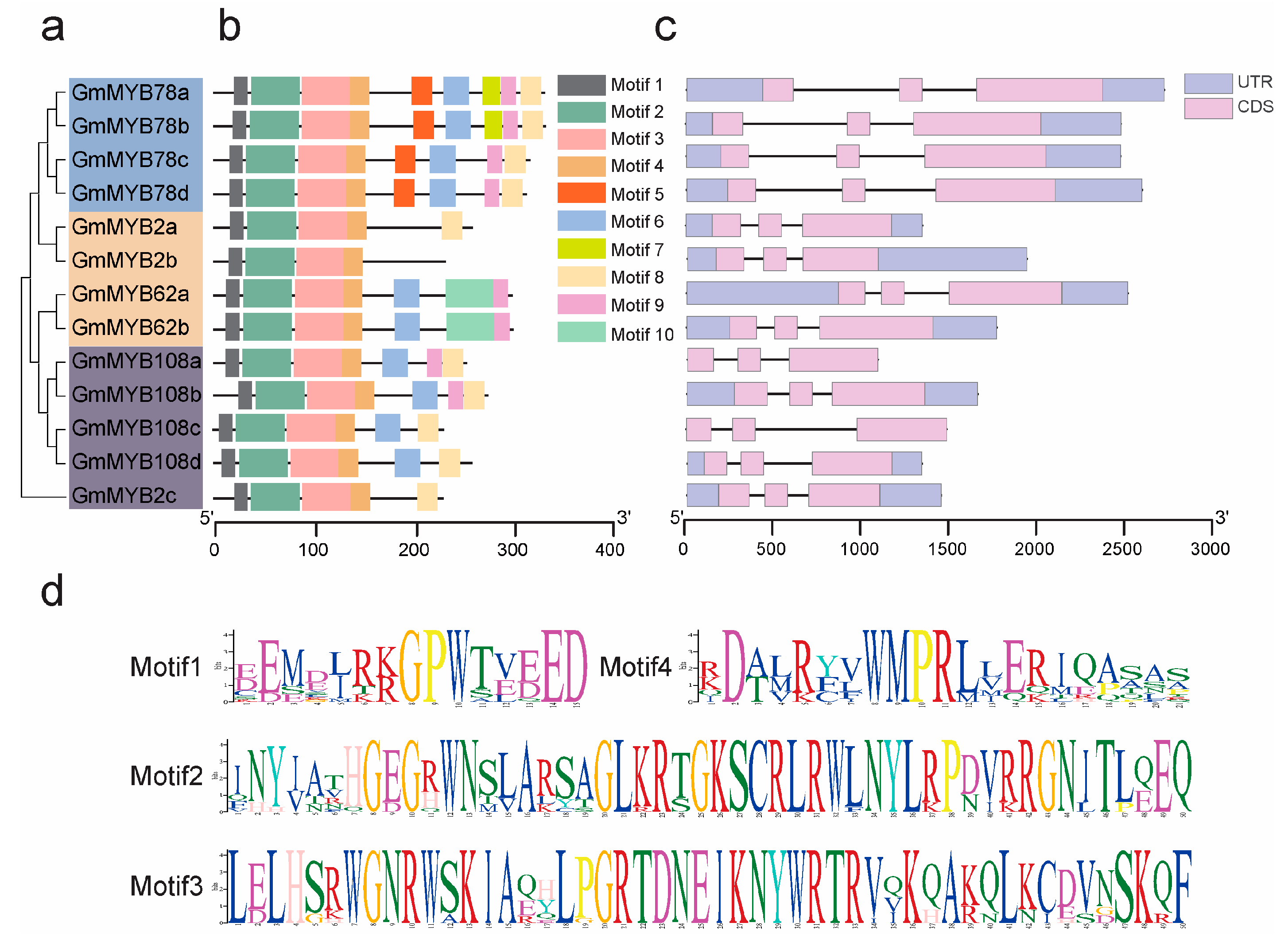
2.3. Structural and Functional Analysis of GmMYB S20
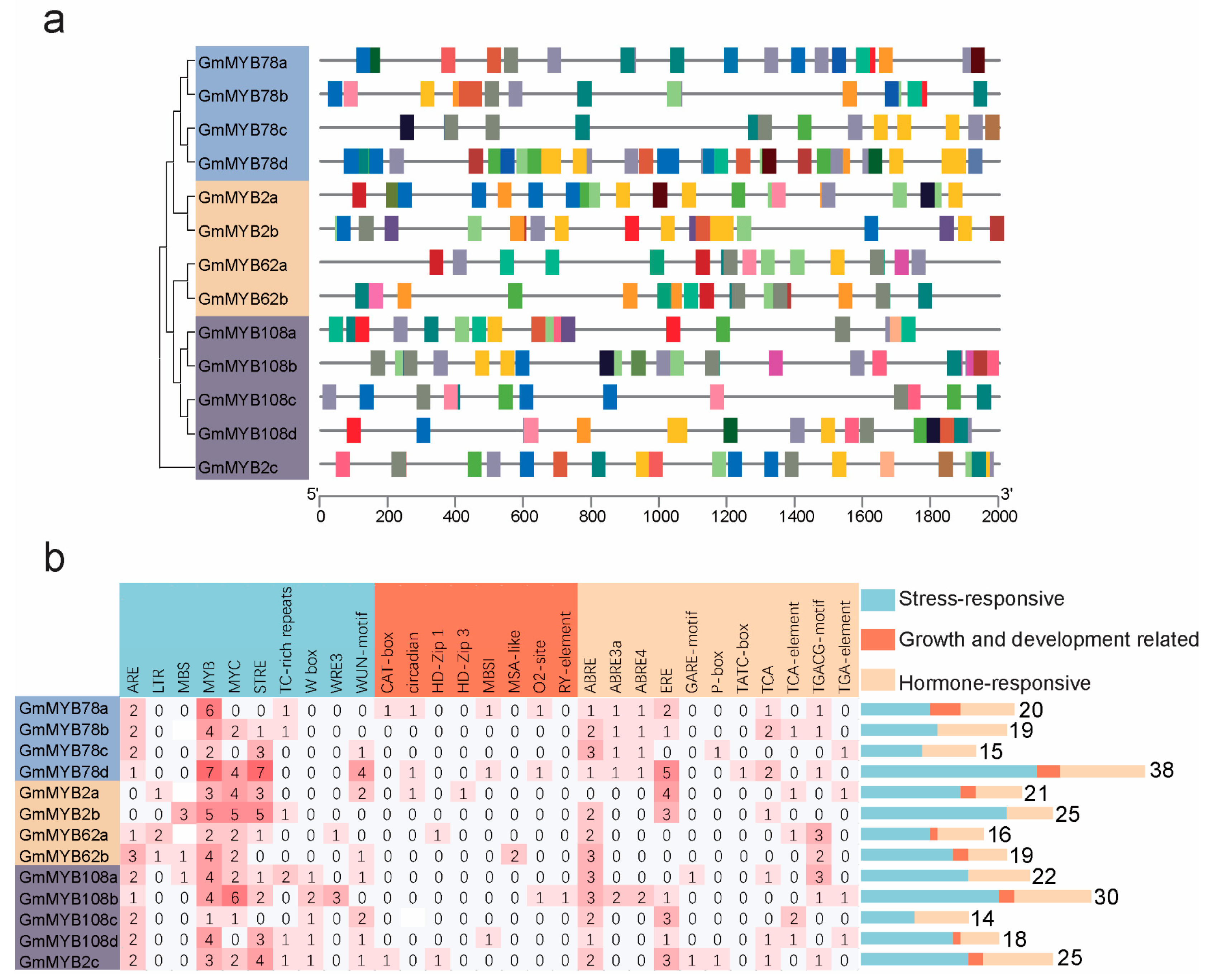
2.4. Cis-Regulatory Elements in GmMYB S20 Promoters
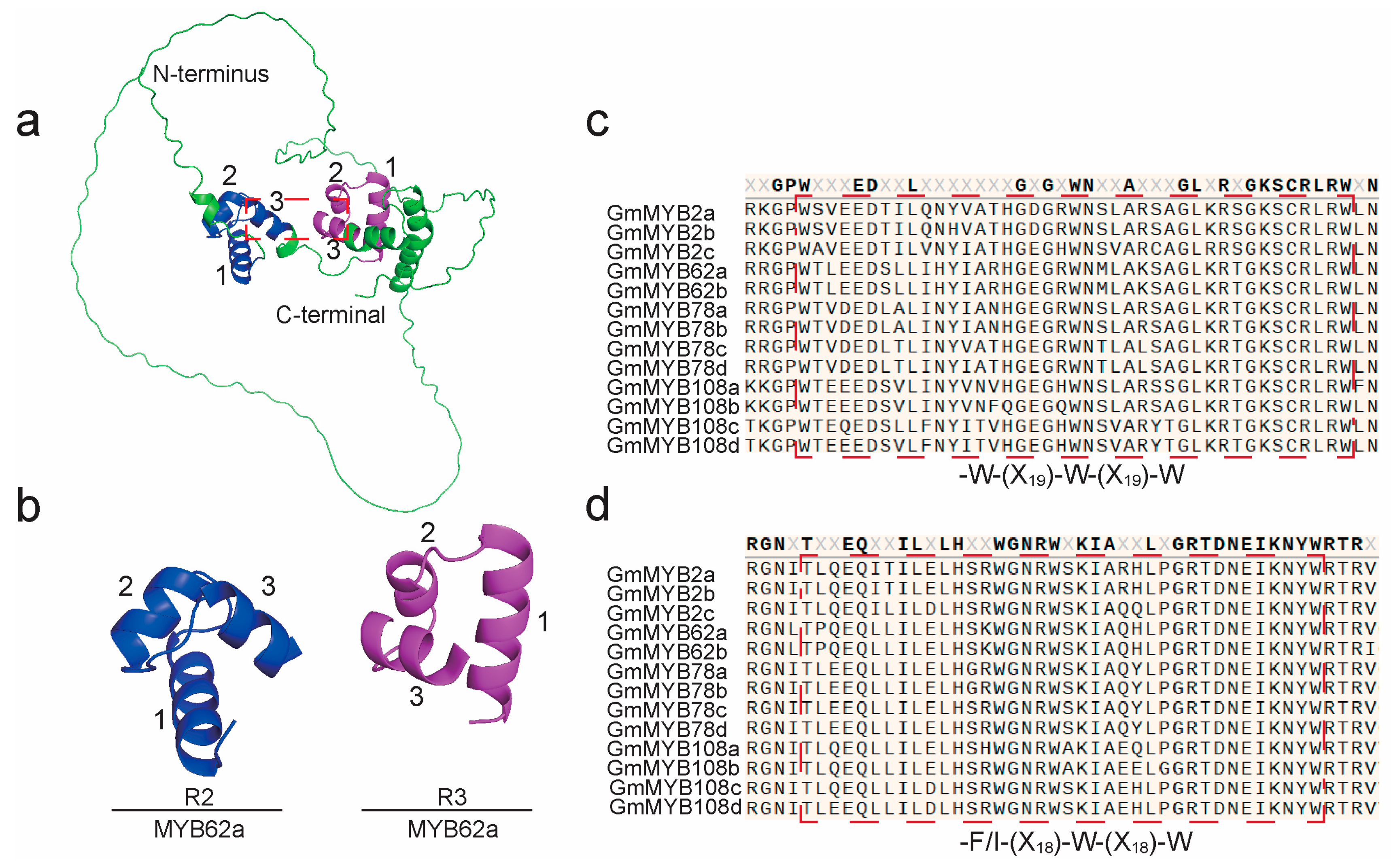
2.5. Structural Characterization of GmMYB S20 Proteins
2.6. GmMYB S20 Genes Expression Patterns
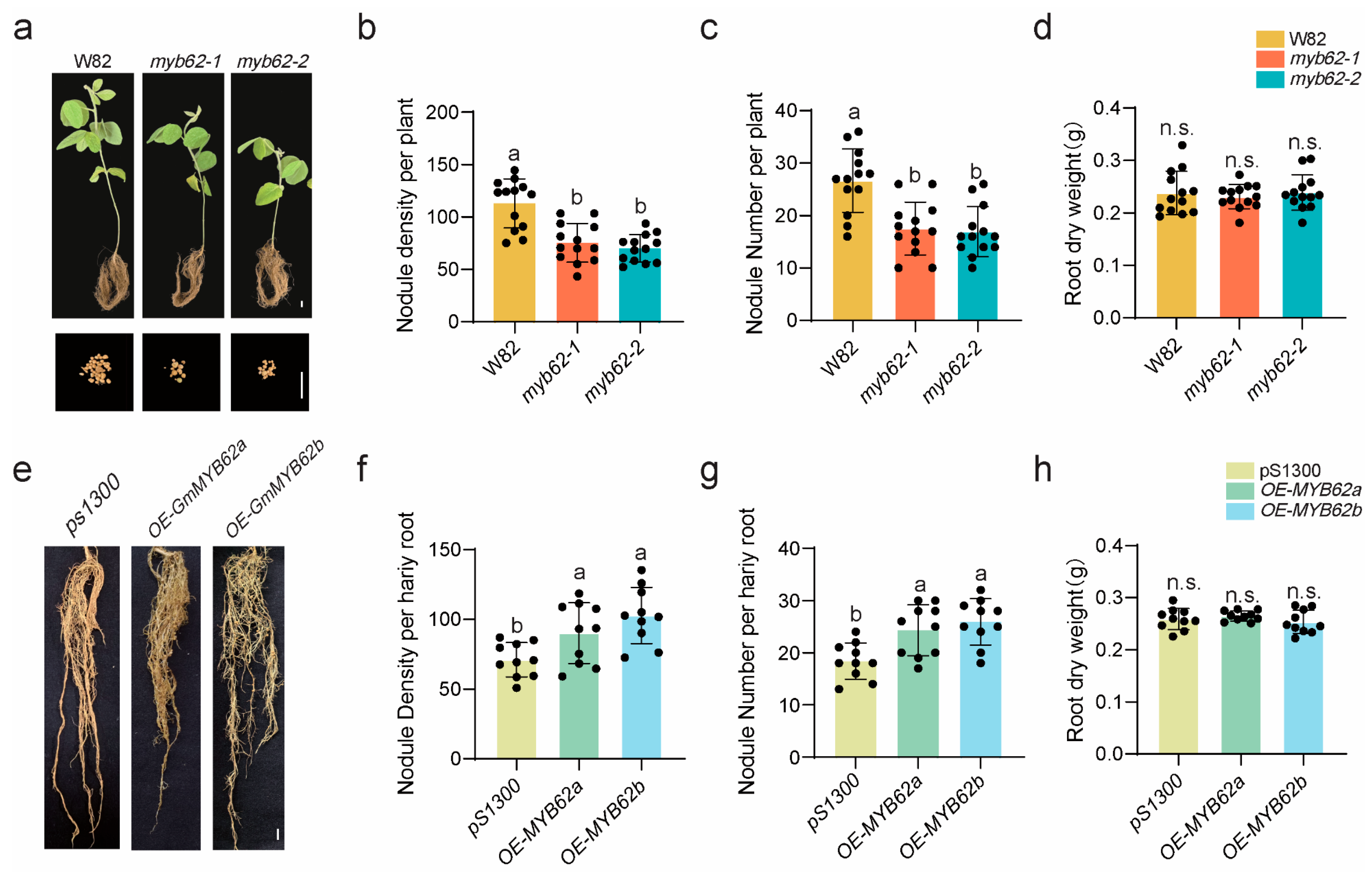
2.7. GmMYB62s Regulate Nodulation
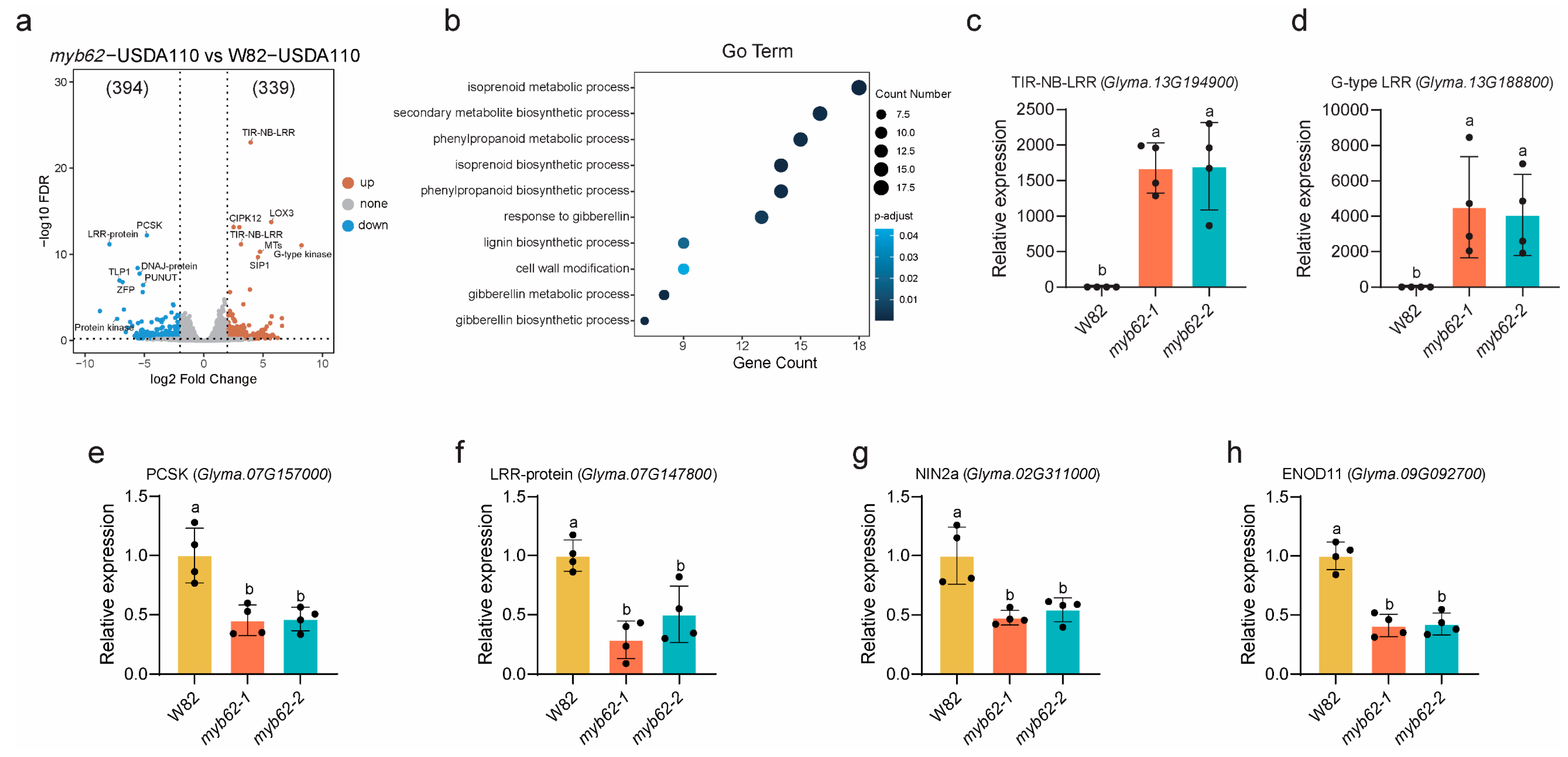
2.8. Transcriptomic Analysis of GmMYB62s Reveals Functional Pathways
3. Discussion
4. Materials and Methods
4.1. Identification and Phylogenetic Analysis of GmMYB S20 Genes
4.2. Chromosomal Localization
4.3. Collinearity Analysis
4.4. Gene Structure and Conserved Motif Analysis
4.5. Prediction of Cis-Regulatory Elements
4.6. Three-Dimensional Protein Structure Modeling
4.7. RNA-Seq Analysis and Functional Enrichment
4.8. Vector Construction
4.9. Plant Materials and Growth Conditions
4.10. Nodule Biomass Measurement
4.11. GUS Histochemical Staining
4.12. Hairy Root Transformation
4.13. Agrobacterium-Mediated Transformation of Soybean
4.14. RNA Extraction and RT-qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, J.H.; Wang, E.T.; Chen, W.F.; Chen, W.X. Genetic diversity and potential for promotion of plant growth detected in nodule endophytic bacteria of soybean grown in Heilongjiang province of China. Soil Biol. Biochem. 2008, 40, 238–246. [Google Scholar] [CrossRef]
- Roy, S.; Liu, W.; Nandety, R.S.; Crook, A.; Mysore, K.S.; Pislariu, C.I.; Frugoli, J.; Dickstein, R.; Udvardi, M.K. Celebrating 20 Years of Genetic Discoveries in Legume Nodulation and Symbiotic Nitrogen Fixation. Plant Cell 2020, 32, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Lam, H.-M.; Nguyen, H.T.; Siddique, K.H.M.; Varshney, R.K.; Colmer, T.D.; Cowling, W.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef] [PubMed]
- Cernay, C.; Ben-Ari, T.; Pelzer, E.; Meynard, J.-M.; Makowski, D. Estimating variability in grain legume yields across Europe and the Americas. Sci. Rep. 2015, 5, 11171. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Zou, Y.; Chen, L.; Cai, Z.; Zhang, S.; Zhao, F.; Tian, Y.; Jiang, Q.; Ferguson, B.J.; et al. Soybean miR172c Targets the Repressive AP2 Transcription Factor NNC1 to Activate ENOD40 Expression and Regulate Nodule Initiation. Plant Cell 2014, 26, 4782–4801. [Google Scholar] [CrossRef]
- Adema, K.; Kohlen, W. The symbiosome-a transient organelle in evolution. J. Exp. Bot. 2024, 75, 3209–3213. [Google Scholar] [CrossRef]
- Jain, V.; Nainawatee, H.S. Plant Flavonoids: Signals to Legume Nodulation and Soil Microorganisms. J. Plant Biochem. Biotechnol. 2002, 11, 1–10. [Google Scholar] [CrossRef]
- Charpentier, M. Calcium signals in the plant nucleus: Origin and function. J. Exp. Bot. 2018, 69, 4165–4173. [Google Scholar] [CrossRef]
- Xie, F.; Murray, J.D.; Kim, J.; Heckmann, A.B.; Edwards, A.; Oldroyd, G.E.D.; Downie, J.A. Legume pectate lyase required for root infection by rhizobia. Proc. Natl. Acad. Sci. USA 2012, 109, 633–638. [Google Scholar] [CrossRef]
- Su, C.; Zhang, G.; Rodriguez-Franco, M.; Hinnenberg, R.; Wietschorke, J.; Liang, P.; Yang, W.; Uhler, L.; Li, X.; Ott, T. Transcellular progression of infection threads in Medicago truncatula roots is associated with locally confined cell wall modifications. Curr. Biol. 2023, 33, 533–542.E5. [Google Scholar] [CrossRef]
- Liu, C.-W.; Breakspear, A.; Stacey, N.; Findlay, K.; Nakashima, J.; Ramakrishnan, K.; Liu, M.; Xie, F.; Endre, G.; de Carvalho-Niebel, F.; et al. A protein complex required for polar growth of rhizobial infection threads. Nat. Commun. 2019, 10, 2848. [Google Scholar] [CrossRef]
- Liang, P.; Schmitz, C.; Lace, B.; Ditengou, F.A.; Su, C.; Schulze, E.; Knerr, J.; Grosse, R.; Keller, J.; Libourel, C.; et al. Formin-mediated bridging of cell wall, plasma membrane, and cytoskeleton in symbiotic infections of Medicago truncatula. Curr. Biol. 2021, 31, 2712–2719.E5. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Oztas, O.; Zhang, X.; Wu, X.; Stonoha, C.; Wang, E.; Wang, B.; Wang, D. A symbiotic SNARE protein generated by alternative termination of transcription. Nat. Plants. 2016, 2, 15197. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Fedorova, E.E.; Limpens, E.; De Mita, S.; Genre, A.; Bonfante, P.; Bisseling, T. Rhizobium–legume symbiosis shares an exocytotic pathway required for arbuscule formation. Proc. Natl. Acad. Sci. USA 2012, 109, 8316–8321. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Ito, M. Conservation and diversification of three-repeat Myb transcription factors in plants. J. Plant Res. 2005, 118, 61–69. [Google Scholar] [CrossRef]
- Su, C.; Feilong, M.; Jiaoyang, C.; Man, Q.; Qianshu, W.; Zhihuan, T.; Bo, S. Function of R2R3-type Myeloblastosis Transcription Factors in Plants. Rice Sci. 2025, 32, 307–321. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) Function as Transcriptional Activators in Abscisic Acid Signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef]
- Yang, A.; Dai, X.; Zhang, W.-H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.; Ding, T.; Lin, X.; Hu, H.; Ding, Z.; Tian, H. MYB2 and MYB108 regulate lateral root development by interacting with LBD29 in Arabidopsis thaliana. J. Integr. Plant Biol. 2024, 66, 1675–1687. [Google Scholar] [CrossRef]
- Bi, H.; Luang, S.; Li, Y.; Bazanova, N.; Morran, S.; Song, Z.; Perera, M.A.; Hrmova, M.; Borisjuk, N.; Lopato, S. Identification and characterization of wheat drought-responsive MYB transcription factors involved in the regulation of cuticle biosynthesis. J. Exp. Bot. 2016, 67, 5363–5380. [Google Scholar] [CrossRef]
- Shriti, S.; Paul, S.; Das, S. Overexpression of CaMYB78 transcription factor enhances resistance response in chickpea against Fusarium oxysporum and negatively regulates anthocyanin biosynthetic pathway. Protoplasma 2023, 260, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Madhuvanthi, R.; Karthikeyan, A.S.; Raghothama, K.G. Phosphate Starvation Responses and Gibberellic Acid Biosynthesis are Regulated by the MYB62 Transcription Factor in Arabidopsis. Mol. Plant 2009, 2, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Y.; Zhao, J.; Zhen, X.; Guo, C.; Shu, Y. Genome-wide identification and characterization of R2R3-MYB genes in Medicago truncatula. Genet. Mol. Biol. 2019, 42, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-C.; Gong, Y.-H.; Tao, T.; Lu, S.; Zhou, W.-Y.; Xia, H.; Zhang, X.-Y.; Yang, Q.-Q.; Zhang, M.-Q.; Hong, L.-M.; et al. Genome-wide identification of R2R3-MYB transcription factor subfamily genes involved in salt stress in rice (Oryza sativa L.). BMC Genom. 2024, 25, 797. [Google Scholar] [CrossRef]
- Ren, R.; Wang, H.; Guo, C.; Zhang, N.; Zeng, L.; Chen, Y.; Ma, H.; Qi, J. Widespread Whole Genome Duplications Contribute to Genome Complexity and Species Diversity in Angiosperms. Mol. Plant 2018, 11, 414–428. [Google Scholar] [CrossRef]
- Fang, T.; Wang, Y.; Chen, H.; Qu, J.; Xiao, P.; Wang, Y.; Jiang, X.; Li, C.; Liu, J.-H. Genome-wide identification and expression profiles of NAC transcription factors in Poncirus trifoliata reveal their potential roles in cold tolerance. BMC Plant Biol. 2025, 25, 633. [Google Scholar] [CrossRef]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. cis-Regulatory Elements in Plant Development, Adaptation, and Evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef]
- Ogata, K.; Hojo, H.; Aimoto, S.; Nakai, T.; Nakamura, H.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structure of a DNA-binding unit of Myb: A helix-turn-helix-related motif with conserved tryptophans forming a hydrophobic core. Proc. Natl. Acad. Sci. USA 1992, 89, 6428–6432. [Google Scholar] [CrossRef]
- Kelemen, Z.; Sebastian, A.; Xu, W.; Grain, D.; Salsac, F.; Avon, A.; Berger, N.; Tran, J.; Dubreucq, B.; Lurin, C.; et al. Analysis of the DNA-Binding Activities of the Arabidopsis R2R3-MYB Transcription Factor Family by One-Hybrid Experiments in Yeast. PLoS ONE 2015, 10, e0141044. [Google Scholar] [CrossRef]
- Ogata, K.; Kanei-Ishii, C.; Sasaki, M.; Hatanaka, H.; Nagadoi, A.; Enari, M.; Nakamura, H.; Nishimura, Y.; Ishii, S.; Sarai, A. The cavity in the hydrophobic core of Myb DNA-binding domain is reserved for DNA recognition and trans-activation. Nat. Struct. Biol. 1996, 3, 178–187. [Google Scholar] [CrossRef]
- Perez-Gil, J.; Behrendorff, J.; Douw, A.; Vickers, C.E. The methylerythritol phosphate pathway as an oxidative stress sense and response system. Nat. Commun. 2024, 15, 5303. [Google Scholar] [CrossRef]
- Fonouni-Farde, C.; Tan, S.; Baudin, M.; Brault, M.; Wen, J.; Mysore, K.S.; Niebel, A.; Frugier, F.; Diet, A. DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection. Nat. Commun. 2016, 7, 12636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, Z.; Su, C.; Wang, Y.; Yan, Q.; Chen, J.; Ott, T.; Li, X. A GmNINa-miR172c-NNC1 Regulatory Network Coordinates the Nodulation and Autoregulation of Nodulation Pathways in Soybean. Mol. Plant 2019, 12, 1211–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Kong, Z. Cellular basis of legume–rhizobium symbiosis. Plant Commun. 2024, 5, 101045. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Praslickova, D.; Ilangumaran, G. Inter-organismal signaling and management of the phytomicrobiome. Front. Plant Sci. 2015, 6, 722. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Austin, M.B.; Stewart, C.; Noel, J.P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef]
- Germain, H.; Séguin, A. Innate immunity: Has poplar made its BED? New Phytol. 2011, 189, 678–687. [Google Scholar] [CrossRef]
- Sun, Y.; Qiao, Z.; Muchero, W.; Chen, J.G. Lectin Receptor-Like Kinases: The Sensor and Mediator at the Plant Cell Surface. Front. Plant Sci. 2020, 11, 596301. [Google Scholar] [CrossRef]
- Vicente, J.; Cascón, T.; Vicedo, B.; García-Agustín, P.; Hamberg, M.; Castresana, C. Role of 9-Lipoxygenase and α-Dioxygenase Oxylipin Pathways as Modulators of Local and Systemic Defense. Mol. Plant 2012, 5, 914–928. [Google Scholar] [CrossRef]
- Fu, M.; Sun, J.; Li, X.; Guan, Y.; Xie, F. Asymmetric redundancy of soybean Nodule Inception (NIN) genes in root nodule symbiosis. Plant Physiol. 2022, 188, 477–489. [Google Scholar] [CrossRef]
- Cerri, M.R.; Frances, L.; Laloum, T.; Auriac, M.C.; Niebel, A.; Oldroyd, G.E.; Barker, D.G.; Fournier, J.; de Carvalho-Niebel, F. Medicago truncatula ERN transcription factors: Regulatory interplay with NSP1/NSP2 GRAS factors and expression dynamics throughout rhizobial infection. Plant Physiol. 2012, 160, 2155–2172. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Lv, A.; Wen, W.; Fan, N.; You, X.; Gao, L.; Zhou, P.; Shi, F.; An, Y. MsMYB206-MsMYB450-MsHY5 complex regulates alfalfa tolerance to salt stress via regulating flavonoid biosynthesis during the day and night cycles. Plant J. 2025, 121, e17216. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Zhou, X.H.; Bao, A.K. Genome-wide analysis of the R2R3-MYB gene family and identification of candidate genes that regulate isoflavone biosynthesis in red clover (Trifolium pratense). Int. J. Biol. Macromol. 2024, 282, 137182. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Chen, Z.; Liu, Q.; Mao, W.; Chen, Y.; Tian, W.; Liu, Y.; Han, J.; Ouyang, X.; Huang, X. Coordinated Transcriptional Regulation by the UV-B Photoreceptor and Multiple Transcription Factors for Plant UV-B Responses. Mol. Plant 2020, 13, 777–792. [Google Scholar] [CrossRef]
- He, Z.; Qin, X.; Jia, T.; Qi, T.; Zhou, Q.; Liu, J.; Peng, Y. Genome-wide identification of 1R-MYB transcription factors family and functional characterization of TrMYB130 under drought stresses in Trifolium repens (L.). Gene 2025, 943, 149247. [Google Scholar] [CrossRef]
- Velandia, K.; Reid, J.B.; Foo, E. Right time, right place: The dynamic role of hormones in rhizobial infection and nodulation of legumes. Plant Commun. 2022, 3, 100327. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Youles, M.; Gruetzner, R.; Ehnert, T.M.; Werner, S.; Jones, J.D.; Patron, N.J.; Marillonnet, S. A golden gate modular cloning toolbox for plants. ACS Synth. Biol. 2014, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Wu, J.; VanDusen, N.J.; Li, Y.; Zheng, Y. CRISPR-Cas9-mediated homology-directed repair for precise gene editing. Mol. Ther. Nucleic Acids 2024, 35, 102344. [Google Scholar] [CrossRef]
- Jefferson, R.A.; Kavanagh, T.A.; Bevan, M.W. GUS fusions: Beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987, 6, 3901–3907. [Google Scholar] [CrossRef]
- Jiaxin Chen, H.M.C.H.Z.L.Y.Q.D.L.B.S.X.L.H.L. A Highly Efficient Method to Generate Chimeric Soybean Plant with Transgenic Hairy Roots. Chin. Bull. Bot. 2024, 59, 89–98. [Google Scholar]
- Cheng, Y.; Wang, X.; Cao, L.; Ji, J.; Liu, T.; Duan, K. Highly efficient Agrobacterium rhizogenes-mediated hairy root transformation for gene functional and gene editing analysis in soybean. Plant Methods 2021, 17, 73. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leng, J.; Xu, R.; Liu, Y.; Jiang, T.; Hu, H.; Ding, Z.; Dai, S. Genome-Wide Analysis of GmMYB S20 Transcription Factors Reveals Their Critical Role in Soybean Nodulation. Plants 2025, 14, 2240. https://doi.org/10.3390/plants14142240
Leng J, Xu R, Liu Y, Jiang T, Hu H, Ding Z, Dai S. Genome-Wide Analysis of GmMYB S20 Transcription Factors Reveals Their Critical Role in Soybean Nodulation. Plants. 2025; 14(14):2240. https://doi.org/10.3390/plants14142240
Chicago/Turabian StyleLeng, Junchen, Ruobing Xu, Yanshuang Liu, Tianshu Jiang, Haiying Hu, Zhaojun Ding, and Shaojun Dai. 2025. "Genome-Wide Analysis of GmMYB S20 Transcription Factors Reveals Their Critical Role in Soybean Nodulation" Plants 14, no. 14: 2240. https://doi.org/10.3390/plants14142240
APA StyleLeng, J., Xu, R., Liu, Y., Jiang, T., Hu, H., Ding, Z., & Dai, S. (2025). Genome-Wide Analysis of GmMYB S20 Transcription Factors Reveals Their Critical Role in Soybean Nodulation. Plants, 14(14), 2240. https://doi.org/10.3390/plants14142240