
Transcriptome and Coexpression Network Analyses Provide Insights into the Resistance of Chinese Cabbage During Different Stages of Plasmodiophora brassicae Infection

Abstract
1. Introduction
2. Results
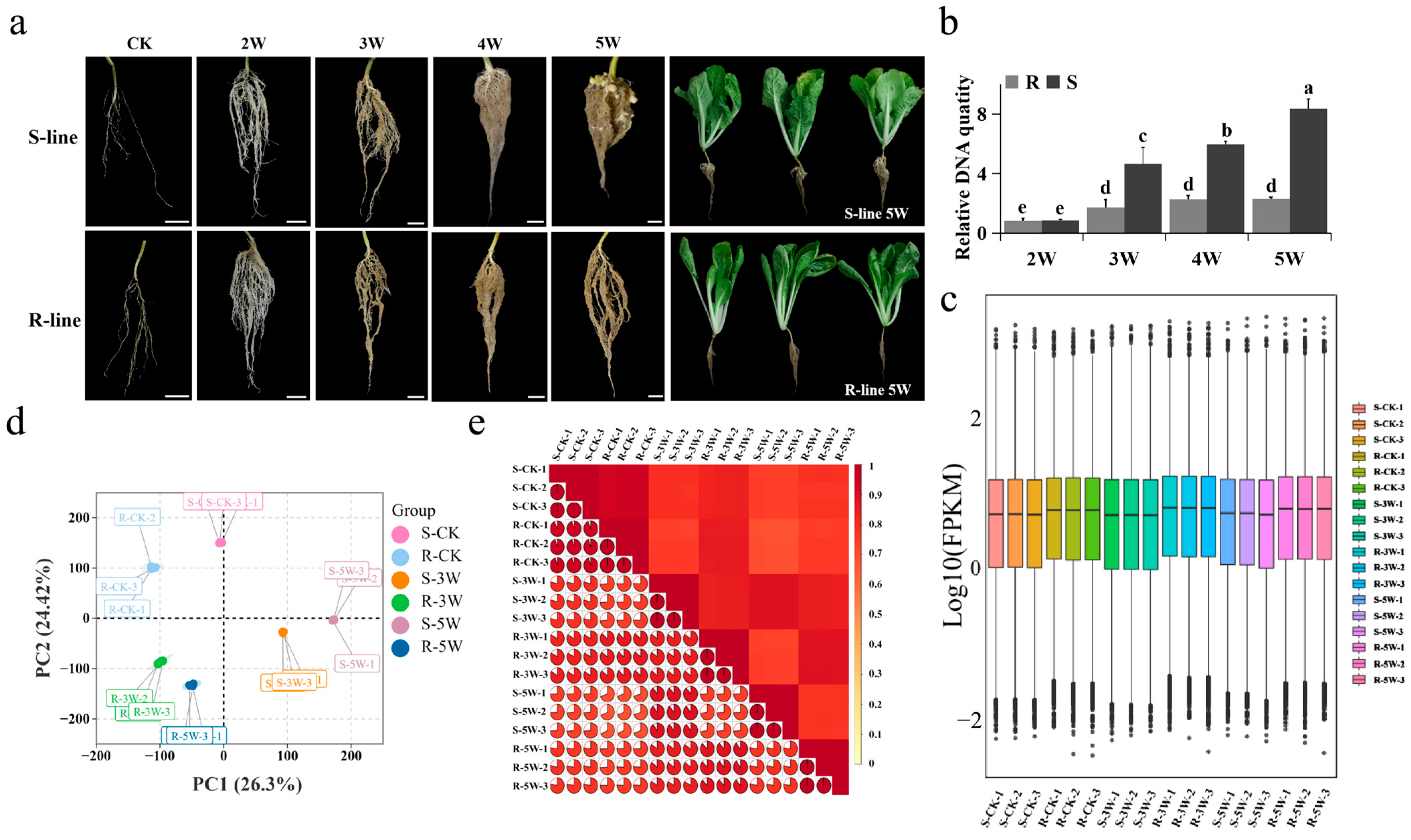
2.1. Phenotypic Observations and Transcriptome Overview
2.2. DEGs in the R- and S-Lines at Different Time Points
2.3. DEG Coexpression Clusters and Functional Analysis
2.4. Identification of Key Genes Responding to P. brassicae Inoculation
3. Discussion
4. Materials and Methods
4.1. Plant Materials and P. brassicae Inoculation
4.2. RNA Extraction, Library Preparation and Transcriptome Sequencing
4.3. DEG Expression Analysis and Functional Annotation
4.4. WGCNA
4.5. Quantification of Gene Expression and RNA-Seq Data Evaluation
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vañó, M.S.; Nourimand, M.; MacLean, A.; Pérez-López, E. Getting to the root of a club-understanding developmental manipulation by the clubroot pathogen. Semin. Cell Dev. Biol. 2023, 148–149, 22–32. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Du, C.Y.; Li, Y.W.; Wang, H.Y.; Zhang, C.Y.; Chen, P. Advances in biological control and resistance genes of Brassicaceae clubroot disease—The Study Case of China. Int. J. Mol. Sci. 2023, 24, 785. [Google Scholar] [CrossRef] [PubMed]
- Dixon, G.R. The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J. Plant Growth Regul. 2009, 28, 194–202. [Google Scholar] [CrossRef]
- Zhu, M.L.; He, Y.W.; Li, Y.; Ren, T.R.; Liu, H.; Huang, J.B.; Jiang, D.H.; Hsiang, T.; Zheng, L. Two new biocontrol agents against clubroot caused by Plasmodiophora brassicae. Front. Microbiol. 2020, 10, 3099. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytol. 2014, 203, 32–43. [Google Scholar] [CrossRef]
- Li, N.Y.; Zhou, L.; Zhang, D.D.; Klosterman, S.J.; Li, T.G.; Gui, Y.J.; Kong, Z.Q.; Ma, X.F.; Short, D.P.G.; Zhang, W.Q.; et al. Heterologous expression of the Cotton NBS-LRR gene GbaNA1 enhances Verticillium Wilt resistance in Arabidopsis. Front. Plant Sci. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Winkelmüller, T.M.; Entila, F.; Anver, S.; Piasecka, A.; Song, B.; Dahms, E.; Sakakibara, H.; Gan, X.; Kułak, K.; Sawikowska, A.; et al. Gene expression evolution in pattern-triggered immunity within Arabidopsis thaliana and across Brassicaceae species. Plant Cell 2021, 33, 1863–1887. [Google Scholar] [CrossRef]
- Hayashi, K.; Fujita, Y.; Ashizawa, T.; Suzuki, F.; Nagamura, Y.; Hayano-Saito, Y. Serotonin attenuates biotic stress and leads to lesion browning caused by a hypersensitive response to Magnaporthe oryzae penetration in rice. Plant J. 2016, 85, 46–56. [Google Scholar] [CrossRef]
- Klessig, D.F.; Choi, H.W.; Dempsey, D.A. Systemic acquired resistance and salicylic acid: Past, Present, and Future. Mol. Plant Microbe Interact. 2018, 31, 871–888. [Google Scholar] [CrossRef]
- Caarls, L.; Pieterse, C.M.; Van Wees, S.C. How salicylic acid takes transcriptional control over jasmonic acid signaling. Front. Plant Sci. 2015, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, A.M.; Argueso, C.T. No hormone to rule them all: Interactions of plant hormones during the responses of plants to pathogens. Semin. Cell Dev. Biol. 2016, 56, 174–189. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, F.; Jiang, L.H.; Chen, C.; Wu, L.T.; Liu, Z.B. Different pathogen defense strategies in Arabidopsis: More than Pathogen Recognition. Cells 2018, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Rana, R.S.; Sureja, A.K.; Verma, M.; Arya, L.; Das Munshi, A. Cloning and characterization of NBS-LRR encoding resistance gene candidates from tomato leaf curl new Delhi virus resistant genotype of Luffa cylindrica Roem. Physiol. Mol. Plant Pathol. 2013, 81, 107–117. [Google Scholar] [CrossRef]
- Couto, D.; Zipfel, C. Regulation of pattern recognition receptor signalling in plants. Nat. Rev. Immunol. 2016, 16, 537–552. [Google Scholar] [CrossRef]
- Kushalappa, A.C.; Yogendra, K.N.; Karre, S. Plant Innate Immune Response: Qualitative and Quantitative Resistance. Crit. Rev. Plant Sci. 2016, 35, 38–55. [Google Scholar] [CrossRef]
- Yu, J.; Li, T.Y.; Tian, L.Y.; Tang, C.; Klosterman, S.J.; Tian, C.M.; Wang, Y.L. Two Verticillium dahliae MAPKKKs, VdSsk2 and VdSte11, have distinct roles in pathogenicity, microsclerotial formation, and stress adaptation. mSphere 2019, 4, e00426-19. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef]
- Manna, M.; Thakur, T.; Chirom, O.; Mandlik, R.; Deshmukh, R.; Salvi, P. Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant. 2021, 172, 847–868. [Google Scholar] [CrossRef]
- Chen, S.S.; Tan, S.X.; Jin, Z.L.; Wu, J.D.; Zhao, Y.Y.; Xu, W.J.; Liu, S.J.; Li, Y.; Huang, H.H.; Bao, F.; et al. The transcriptional landscape of Populus pattern/effector-triggered immunity and how PagWRKY18 involved in it. Plant Cell Environ. 2024, 47, 2074–2092. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Kracher, B.; Ross, A.; Kramer, K.; Finkemeier, I.; Somssich, I.E. Principles and characteristics of the Arabidopsis WRKY regulatory network during early MAMP-triggered immunity. Plant J. 2018, 96, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Chhapekar, S.S.; Rameneni, J.J.; Oh, S.H.; Dhandapani, V.; Subburaj, S.; Shin, S.Y.; Ramchiary, N.; Shin, C.; Choi, S.R.; et al. MiR1885 regulates disease tolerance genes in Brassica rapa during early infection with Plasmodiophora brassicae. Int. J. Mol. Sci. 2021, 22, 9433. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhang, J.H.; Deng, C.J.; Yuan, J.S.; Wang, B.W.; Meng, H.; Mohany, M.; Zeng, L.T.; Wei, L.F.; Ahmed, W.; et al. Comparative transcriptome analysis reveals molecular mechanisms of resistance in Chinese cabbage to Plasmodiophora brassicae pathotype 11. Front. Microbiol. 2025, 16, 1495243. [Google Scholar] [CrossRef]
- Meng, S.D.; Yan, X.Y.; Piao, Y.L.; Li, S.Z.; Wang, X.; Jiang, J.; Liang, Y.; Pang, W.X. Multiple transcription factors involved in the response of Chinese cabbage against Plasmodiophora brassicae. Front. Plant Sci. 2024, 15, 1391173. [Google Scholar] [CrossRef]
- Miyaji, N.; Akter, M.A.; Shimizu, M.; Mehraj, H.; Doullah, M.A.; Dennis, E.S.; Chuma, I.; Fujimoto, R. Differences in the transcriptional immune response to Albugo candida between white rust resistant and susceptible cultivars in Brassica rapa L. Sci. Rep. 2023, 13, 8599. [Google Scholar] [CrossRef]
- Antanynienė, R.; Kurgonaitė, M.; Mažeikienė, I.; Frercks, B. Time-Series Transcriptome Analysis of the European Plum Response to Pathogen Monilinia fructigena. Agriculture 2025, 15, 788. [Google Scholar] [CrossRef]
- Geng, D.L.; Shen, X.X.; Xie, Y.P.; Yang, Y.S.; Bian, R.L.; Gao, Y.Q.; Li, P.M.; Sun, L.Y.; Feng, H.; Ma, F.W.; et al. Regulation of phenylpropanoid biosynthesis by MdMYB88 and MdMYB124 contributes to pathogen and drought resistance in apple. Hortic. Res. 2020, 7, 102. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Cao, G.Z.; Li, X.N.; Piao, Z.Y. Effects of exogenous Ergothioneine on Brassica rapa clubroot development revealed by transcriptomic analysis. Int. J. Mol. Sci. 2023, 24, 6380. [Google Scholar] [CrossRef]
- Chen, J.J.; Pang, W.X.; Chen, B.; Zhang, C.Y.; Piao, Z.Y. Transcriptome analysis of Brassica rapa near-isogenic lines carrying clubroot-resistant and -susceptible alleles in response to Plasmodiophora brassicae during early infection. Front. Plant Sci. 2016, 6, 1183. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Zhang, J.; Wang, Y.L.; Fang, B.; Ge, W.J.; Wang, X.L.; Zou, J.W.; Ji, R.Q. Transcriptome analysis of Chinese Cabbage infected with Plasmodiophora Brassicae in the primary stage. Sci. Rep. 2024, 14, 26180. [Google Scholar] [CrossRef] [PubMed]
- Sekhwal, M.K.; Li, P.; Lam, I.; Wang, X.; Cloutier, S.; You, F.M. Disease resistance gene analogs (RGAs) in Plants. Int. J. Mol. Sci. 2015, 16, 19248–19290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, J.L.; Tong, L.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Zhang, Y.Y.; Lv, H.H. Global survey of the full-length Cabbage transcriptome (Brassica oleracea Var. capitata L.) reveals key alternative splicing events involved in growth and disease response. Int. J. Mol. Sci. 2021, 22, 10443. [Google Scholar]
- Zhao, Y.; Bi, K.; Gao, Z.X.; Chen, T.; Liu, H.Q.; Xie, J.T.; Cheng, J.S.; Fu, Y.P.; Jiang, D.H. Transcriptome analysis of Arabidopsis thaliana in response to Plasmodiophora brassicae during early infection. Front. Microbiol. 2017, 8, 673. [Google Scholar] [CrossRef]
- Yang, Y.X.; Ahammed, G.J.; Wu, C.; Fan, S.Y.; Zhou, Y.H. Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr. Protein Pept. Sci. 2015, 16, 450–461. [Google Scholar] [CrossRef]
- Nomoto, M.; Skelly, M.J.; Itaya, T.; Mori, T.; Suzuki, T.; Matsushita, T.; Tokizawa, M.; Kuwata, K.; Mori, H.; Yamamoto, Y.Y.; et al. Suppression of MYC transcription activators by the immune cofactor NPR1 fine-tunes plant immune responses. Cell Rep. 2021, 37, 110125. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Song, W.; Yang, Z.; Li, L.; Ma, Q.; Tan, X.; Wei, Z.; Li, Y.; Li, J.; et al. Different viral effectors suppress hormone-mediated antiviral immunity of rice coordinated by OsNPR1. Nat. Commun. 2023, 14, 3011. [Google Scholar] [CrossRef]
- Lorenzo, O.; Chico, J.M.; Sánchez-Serrano, J.J.; Solano, R. JASMONATE-INSENSITIVE1 encodes a MYC transcription factor essential to discriminate between different jasmonate-regulated defense responses in Arabidopsis. Plant Cell 2004, 16, 1938–1950. [Google Scholar] [CrossRef]
- Song, S.; Huang, H.; Gao, H.; Wang, J.; Wu, D.; Liu, X.; Yang, S.; Zhai, Q.; Li, C.; Qi, T.; et al. Interaction between MYC2 and ETHYLENE INSENSITIVE3 modulates antagonism between jasmonate and ethylene signaling in Arabidopsis. Plant Cell 2014, 26, 263–279. [Google Scholar] [CrossRef]
- Zhou, Y.B.; Xu, S.C.; Jiang, N.; Zhao, X.H.; Bai, Z.N.; Liu, J.L.; Yao, W.; Tang, Q.Y.; Xiao, G.; Lv, C.; et al. Engineering of rice varieties with enhanced resistances to both blast and bacterial blight diseases via CRISPR/Cas9. Plant Biotechnol. J. 2022, 20, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.C.; Simmons, B.A.; Sale, K.L. Structure-based engineering of a plant-fungal hybrid peroxidase for enhanced temperature and pH tolerance. Cell Chem. Biol. 2018, 25, 974–983.e3. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Bi, K.; He, Z.C.; Gao, Z.X.; Zhao, Y.; Fu, Y.P.; Cheng, J.S.; Xie, J.T.; Jiang, D.H. Arabidopsis mutant bik1 exhibits strong resistance to Plasmodiophora brassicae. Front. Physiol. 2016, 7, 402. [Google Scholar] [CrossRef]
- Weigel, R.R.; Pfitzner, U.M.; Gatz, C. Interaction of NIMIN1 with NPR1 modulates PR gene expression in Arabidopsis. Plant Cell 2005, 17, 1279–1291. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, P.; Li, M. Research progress of peptides discovery and function in resistance to abiotic stress in plant. Stress Biol. 2025, 5, 36. [Google Scholar] [CrossRef]
- Wang, D.C.; Wei, L.R.; Ma, J.B.; Wan, Y.Q.; Huang, K.Y.; Sun, Y.Q.; Wen, H.L.; Chen, Z.P.; Li, Z.J.; Yu, D.L.; et al. Bacillus cereus NJ01 induces SA- and ABA-mediated immunity against bacterial pathogens through the EDS1-WRKY18 module. Cell Rep. 2024, 43, 113985. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.; Töller, A.; Diezel, C.; Roth, C.; Westphal, L.; Wiermer, M.; Somssich, I.E. Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant Microbe Interact. 2013, 26, 758–767. [Google Scholar] [CrossRef]
- Wang, X.; Qiao, Q.H.; Zhao, K.K.; Zhai, W.H.; Zhang, F.; Dong, H.Z.; Lin, L.K.; Xing, C.H.; Su, Z.Y.; Pan, Z.J.; et al. PbWRKY18 promotes resistance against black spot disease by activation of the chalcone synthase gene PbCHS3 in pear. Plant Sci. 2024, 341, 112015. [Google Scholar] [CrossRef]
- Kou, E.F.; Luo, Z.X.; Ye, J.Y.; Chen, X.; Lu, D.; Landry, M.P.; Zhang, H.L.; Zhang, H. Sunlight-sensitive carbon dots for plant immunity priming and pathogen defence. Plant Biotechnol. J. 2025; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.X.; Liang, Y.; Zhan, Z.X.; Li, X.N.; Piao, Z.Y. Development of a sinitic clubroot differential set for the pathotype classification of Plasmodiophora brassicae. Front. Plant Sci. 2020, 11, 568771. [Google Scholar] [CrossRef]
- Liu, H.S.; Nwafor, C.C.; Piao, Y.L.; Li, X.N.; Zhan, Z.X.; Piao, Z.Y. Identification and characterization of circular RNAs in Brassica rapa in response to Plasmodiophora brassicae. Int. J. Mol. Sci. 2022, 23, 5369. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Michaelidou, K.; Tzovaras, A.; Missitzis, I.; Ardavanis, A.; Scorilas, A. The expression of the CEACAM19 gene, a novel member of the CEA family, is associated with breast cancer progression. Int. J. Oncol. 2013, 42, 1770–1777. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | KEGG_ID | Description | p-Value |
|---|---|---|---|
| Midnight blue | ko04626 | Plant–pathogen interaction | 5.36166 × 10−5 |
| ko02010 | ABC transporters | 0.001097744 | |
| ko00908 | Zeatin biosynthesis | 0.011942757 | |
| ko00944 | Flavone and flavonol biosynthesis | 0.03415298 | |
| ko01110 | Biosynthesis of secondary metabolites | 0.034815157 | |
| ko04016 | MAPK signaling pathway–plant | 0.045562111 | |
| green | ko04626 | Plant-pathogen interaction | 5.90315 × 10−14 |
| ko04016 | MAPK signaling pathway-plant | 4.13077 × 10−5 | |
| ko04075 | Plant hormone signal transduction | 7.81607 × 10−5 | |
| ko00073 | Cutin, suberine and wax biosynthesis | 0.012763979 | |
| ko00380 | Tryptophan metabolism | 0.024950856 | |
| ko00562 | Inositol phosphate metabolism | 0.029331177 | |
| ko00601 | Glycosphingolipid biosynthesis–lacto and neolacto series | 0.042280796 | |
| ko00561 | Glycerolipid metabolism | 0.049712822 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Wang, L.; Wang, G.; Wu, H.; Wang, X. Transcriptome and Coexpression Network Analyses Provide Insights into the Resistance of Chinese Cabbage During Different Stages of Plasmodiophora brassicae Infection. Plants 2025, 14, 2105. https://doi.org/10.3390/plants14142105
Liu H, Wang L, Wang G, Wu H, Wang X. Transcriptome and Coexpression Network Analyses Provide Insights into the Resistance of Chinese Cabbage During Different Stages of Plasmodiophora brassicae Infection. Plants. 2025; 14(14):2105. https://doi.org/10.3390/plants14142105
Chicago/Turabian StyleLiu, Huishan, Lili Wang, Guozheng Wang, Haidong Wu, and Xin Wang. 2025. "Transcriptome and Coexpression Network Analyses Provide Insights into the Resistance of Chinese Cabbage During Different Stages of Plasmodiophora brassicae Infection" Plants 14, no. 14: 2105. https://doi.org/10.3390/plants14142105
APA StyleLiu, H., Wang, L., Wang, G., Wu, H., & Wang, X. (2025). Transcriptome and Coexpression Network Analyses Provide Insights into the Resistance of Chinese Cabbage During Different Stages of Plasmodiophora brassicae Infection. Plants, 14(14), 2105. https://doi.org/10.3390/plants14142105