
Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome-Wide Identification and Phylogenetic Analysis of P. euphratica AP Gene Family
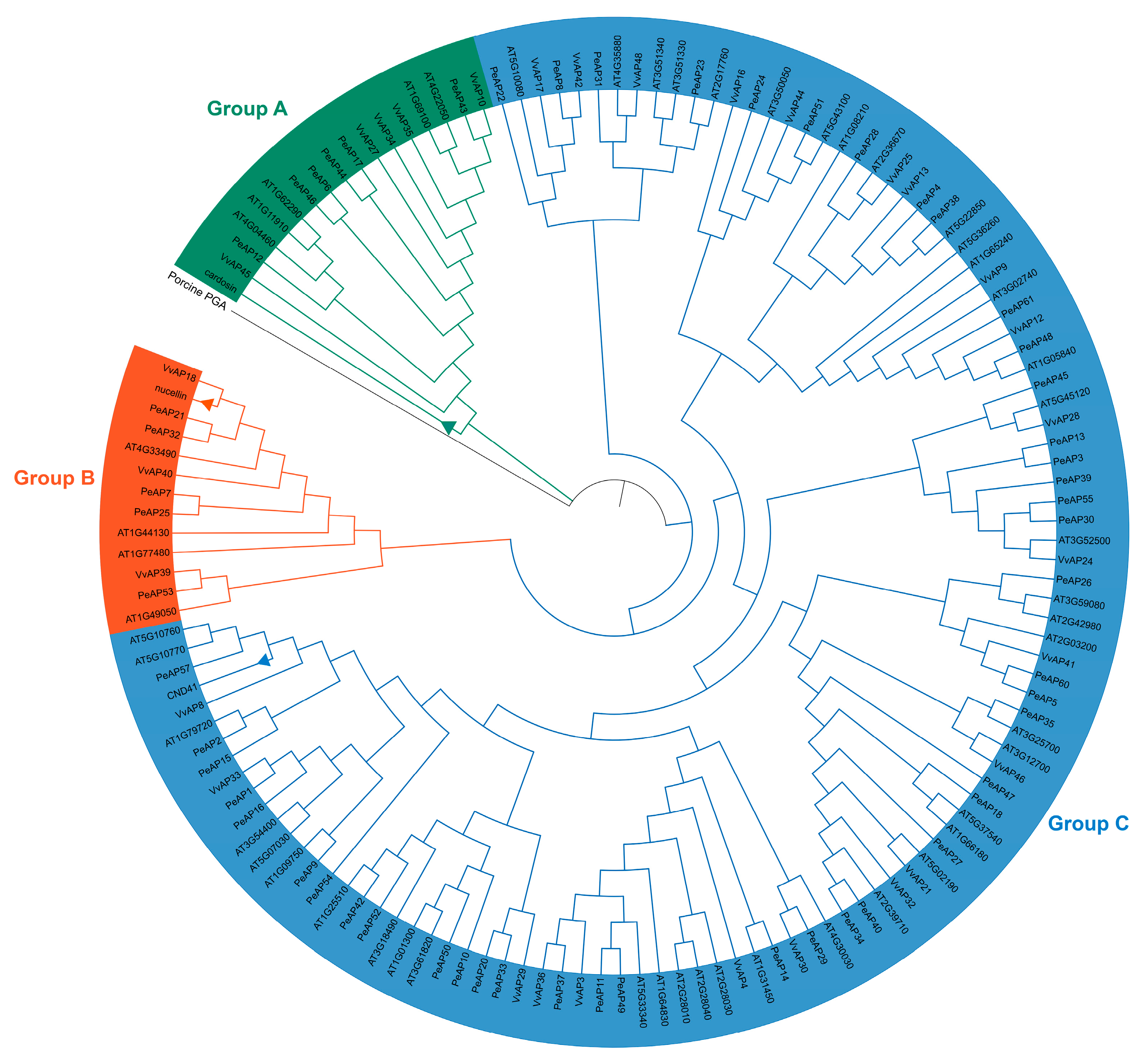
2.2. Phylogenetic Analysis of the PeAP Proteins
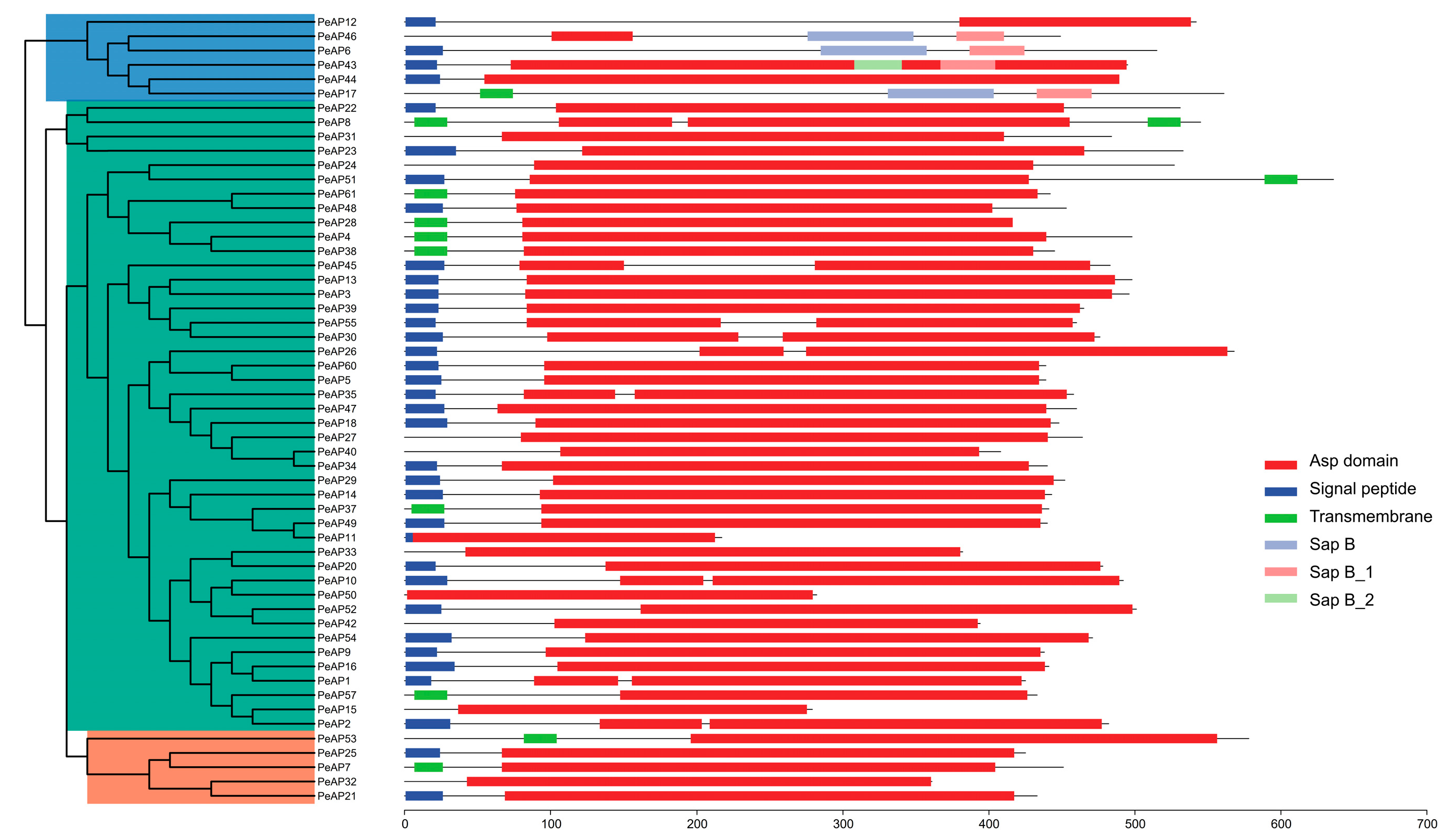
2.3. Predicted Structure and Conserved Motifs Analysis of PeAP Proteins
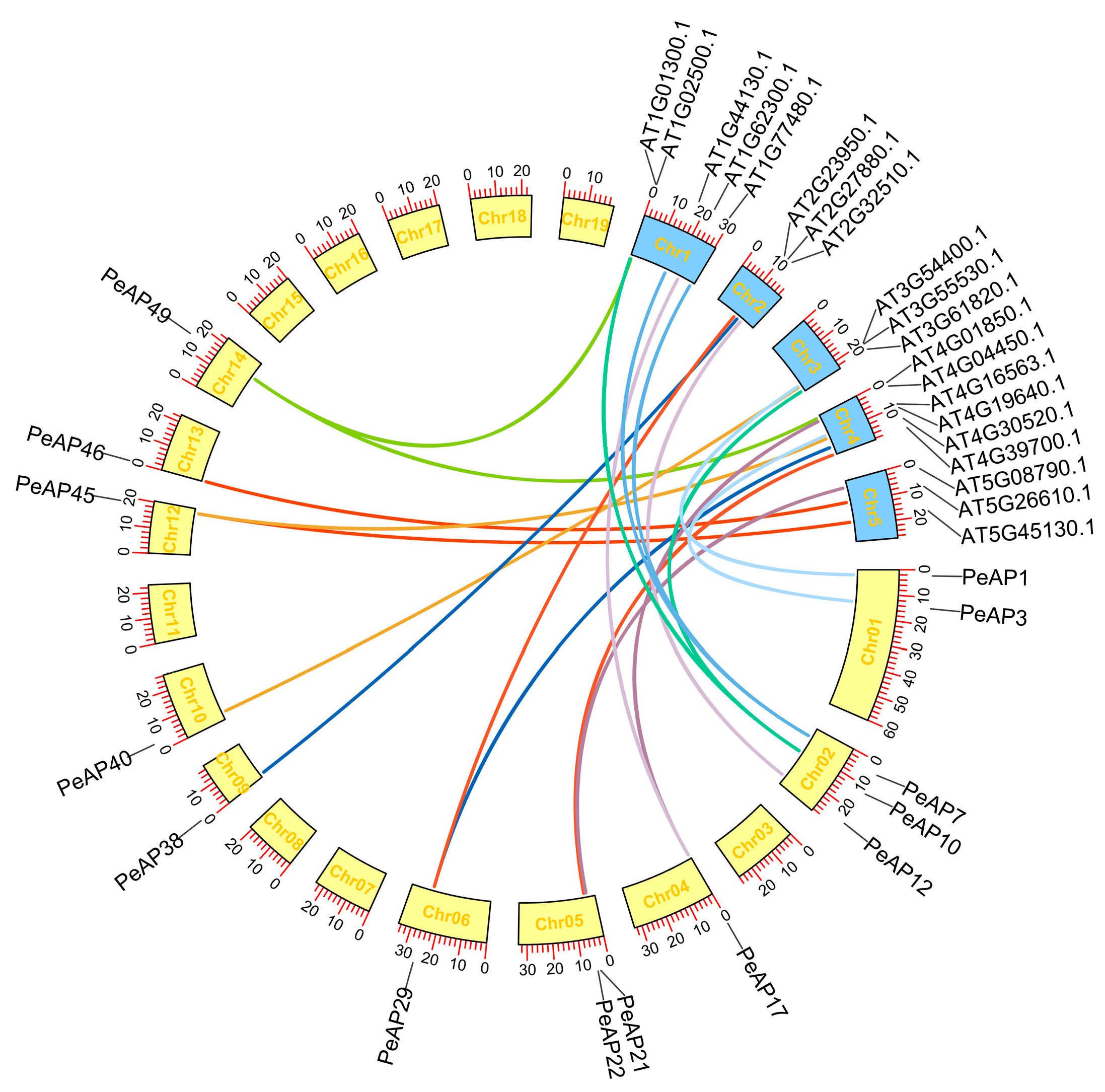
2.4. Evolutionary Relationships of AP Genes Between P. euphratica and Arabidopsis
2.5. Subcellular Localization Prediction of PeAPs
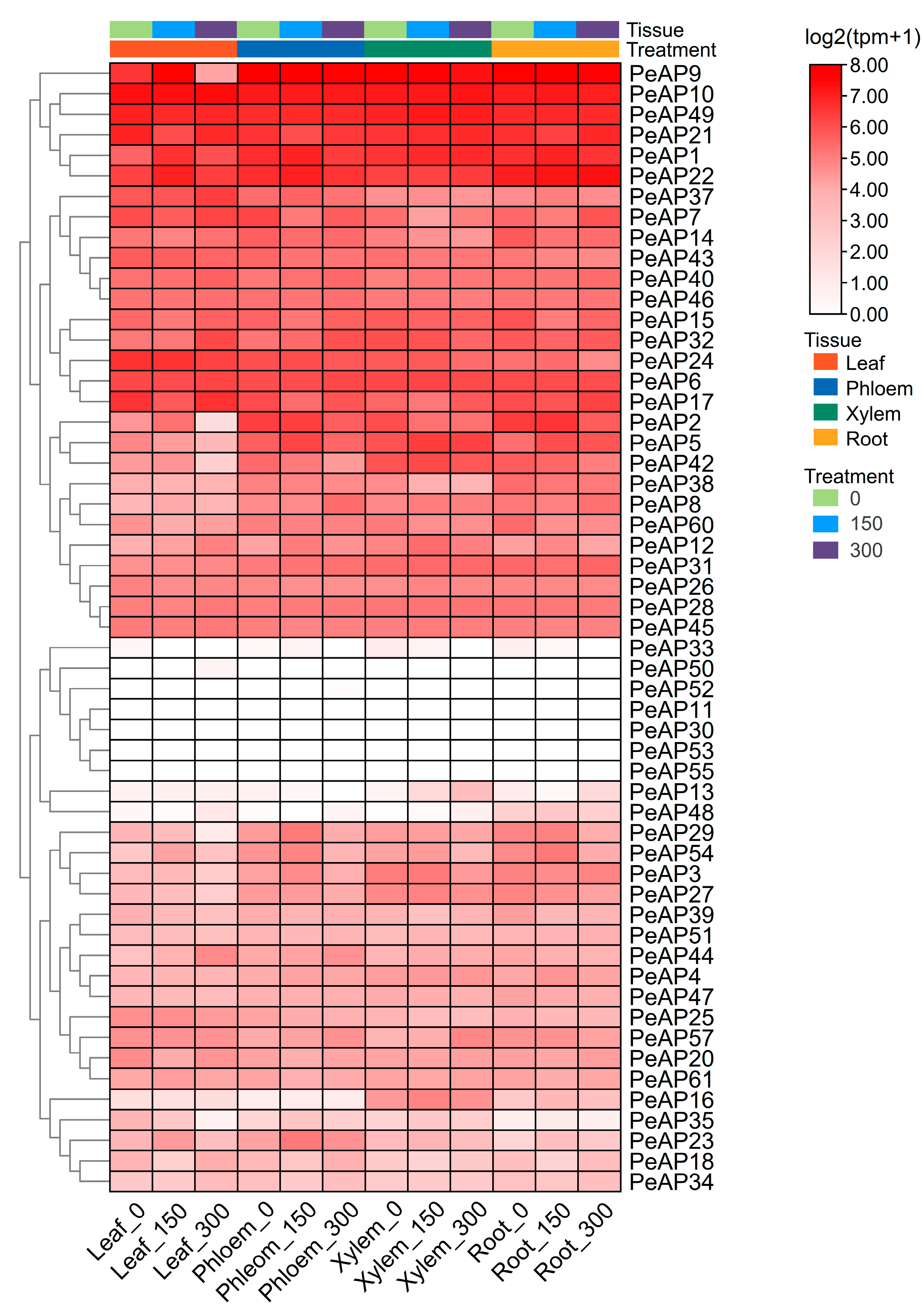
2.6. Spatial and Temporal Expression of PeAPs in Various Developmental Tissues of P. euphratica Under Salt Treatment
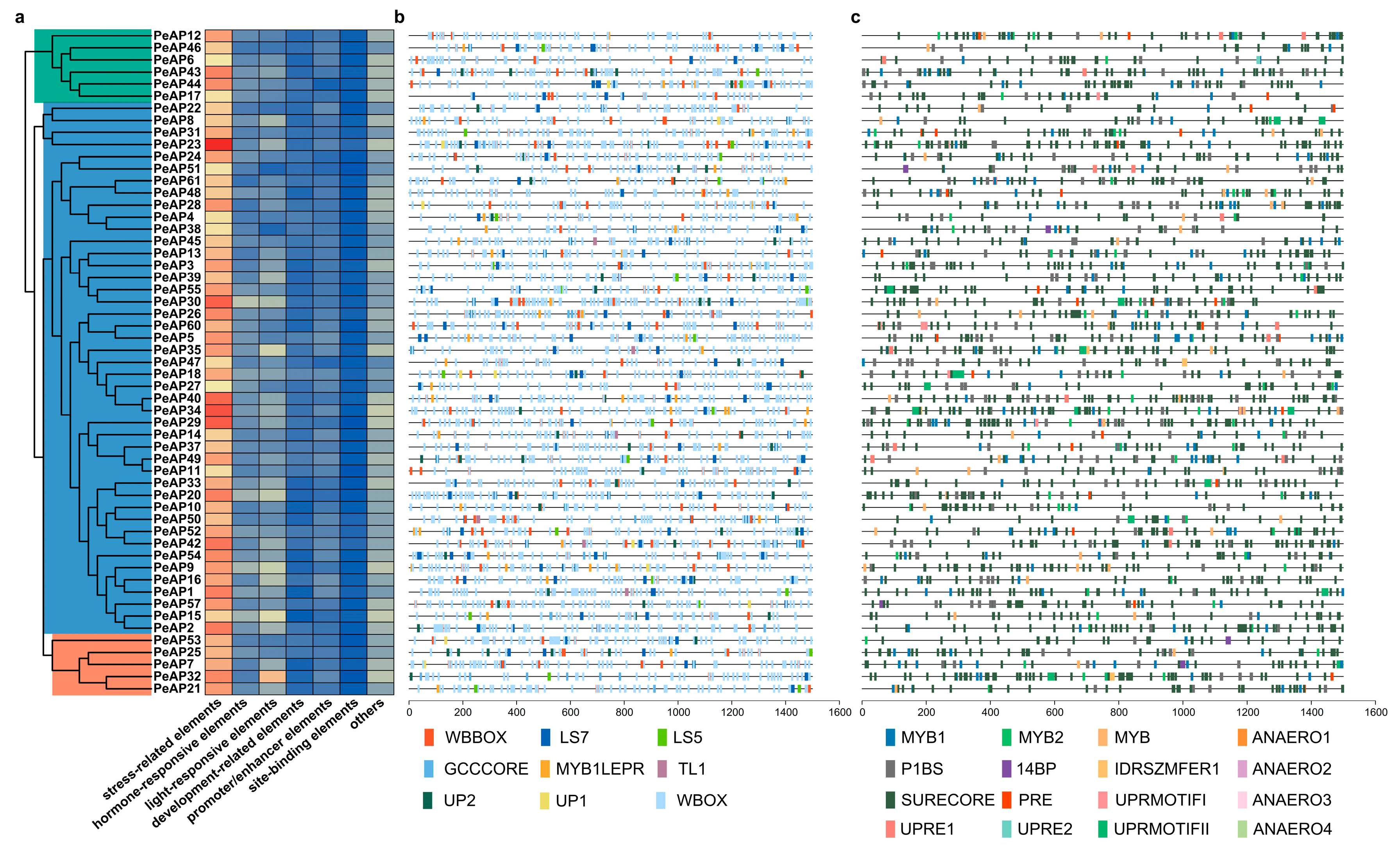
2.7. Prediction of Cis-Acting Elements in the Promoter Regions of the PeAP Genes
2.8. Regulatory Network Mediated by P. euphratica AP Genes
3. Discussion
4. Materials and Methods
4.1. Identification of AP Genes in Populus euphratica
4.2. Multiple Sequence Alignment and Phylogenetic Analysis
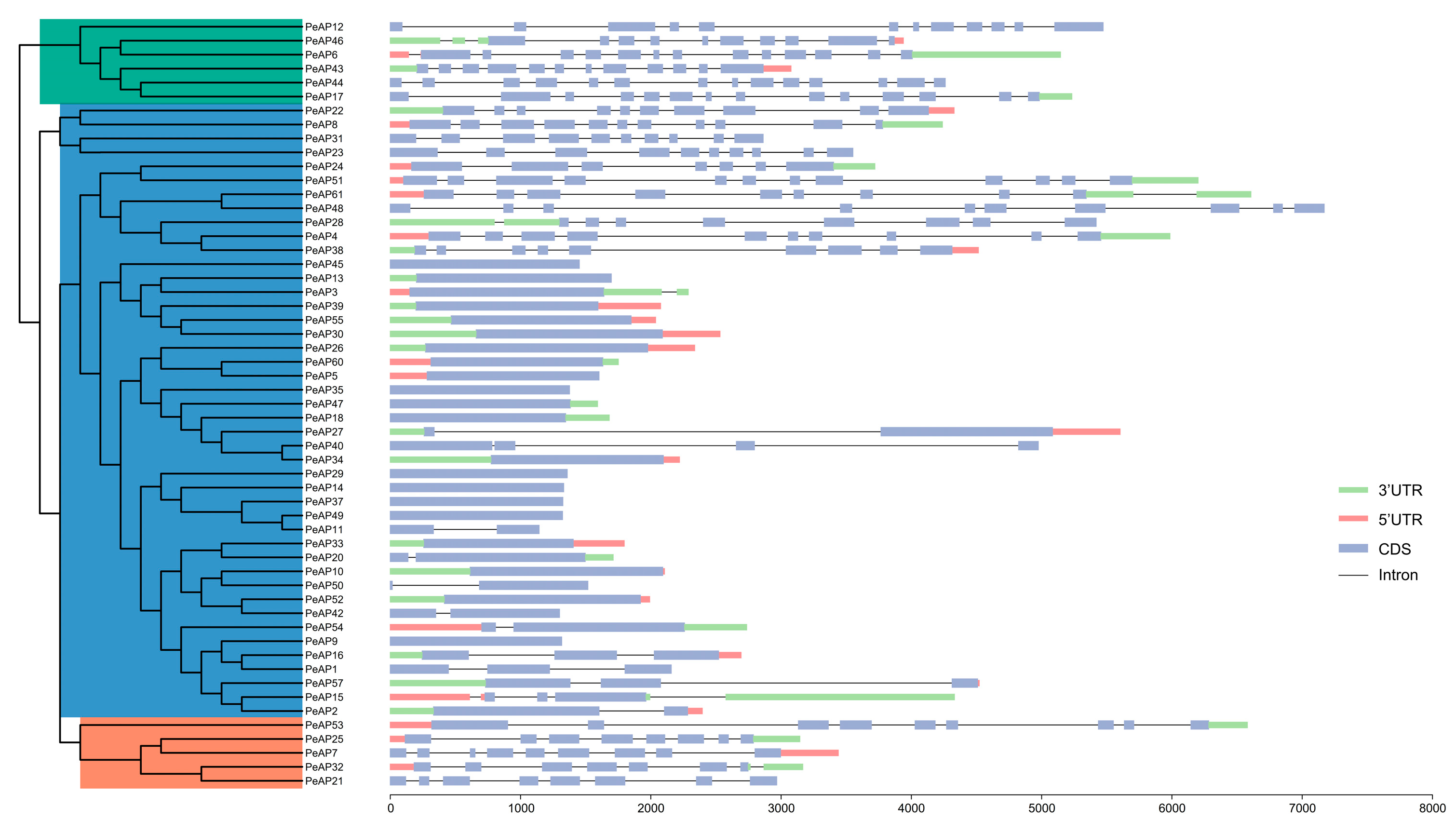
4.3. Gene and Protein Structure Analysis
4.4. Cis-Elements Identification
4.5. Analysis of Transcriptional Profiles
4.6. Analyses of Synteny in PeAPs
4.7. Prediction of 3-Dimensional Structures and Interaction Network of PeAP Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shrivastava, P.; Kumar, R. Soil salinity: A serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation. Saudi J. Biol. Sci. 2015, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Arif, Y.; Singh, P.; Siddiqui, H.; Bajguz, A.; Hayat, S. Salinity induced physiological and biochemical changes in plants: An omic approach towards salt stress tolerance. Plant Physiol. Biochem. 2020, 156, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, T.; Shen, G.; Esmaeili, N.; Zhang, H. Plants’ Response Mechanisms to Salinity Stress. Plants 2023, 12, 2253. [Google Scholar] [CrossRef]
- Chen, S.; Hawighorst, P.; Sun, J.; Polle, A. Salt tolerance in Populus: Significance of stress signaling networks, mycorrhization, and soil amendments for cellular and whole-plant nutrition. Environ. Exp. Bot. 2014, 107, 113–124. [Google Scholar] [CrossRef]
- Watanabe, S.; Kojima, K.; Ide, Y.; Sasaki, S. Effects of saline and osmotic stress on proline and sugar accumulation in Populus euphratica in vitro. Plant Cell Tissue Organ Cult. (PCTOC) 2000, 63, 199–206. [Google Scholar] [CrossRef]
- Soares, A.; Ribeiro Carlton, S.M.; Simões, I. Atypical and nucellin-like aspartic proteases: Emerging players in plant developmental processes and stress responses. J. Exp. Bot. 2019, 70, 2059–2076. [Google Scholar] [CrossRef]
- Cao, S.; Guo, M.; Wang, C.; Xu, W.; Shi, T.; Tong, G.; Zhen, C.; Cheng, H.; Yang, C.; Elsheery, N.I.; et al. Genome-wide characterization of aspartic protease (AP) gene family in Populus trichocarpa and identification of the potential PtAPs involved in wood formation. BMC Plant Biol. 2019, 19, 276. [Google Scholar] [CrossRef]
- Simoes, I.; Faro, C. Structure and function of plant aspartic proteinases. Eur. J. Biochem. 2004, 271, 2067–2075. [Google Scholar] [CrossRef]
- Barrett, A.J. Cellular Proteolysis an Overview. Ann. New York Acad. Sci. 1992, 674, 1–15. [Google Scholar] [CrossRef]
- Faro, C.; Gal, S. Aspartic proteinase content of the Arabidopsis genome. Curr. Protein Pept. Sci. 2005, 6, 493–500. [Google Scholar] [CrossRef]
- Chen, J.; Ouyang, Y.; Wang, L.; Xie, W.; Zhang, Q. Aspartic proteases gene family in rice: Gene structure and expression, predicted protein features and phylogenetic relation. Gene 2009, 442, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Tu, M.; Wang, X.; Zhao, J.; Wan, R.; Li, Z.; Wang, Y.; Wang, X. Ectopic expression of a grape aspartic protease gene, AP13, in Arabidopsis thaliana improves resistance to powdery mildew but increases susceptibility to Botrytis cinerea. Plant Sci. 2016, 248, 17–27. [Google Scholar] [CrossRef]
- Yao, X.; Xiong, W.; Ye, T.; Wu, Y. Overexpression of the aspartic protease ASPG1 gene confers drought avoidance in Arabidopsis. J. Exp. Bot. 2012, 63, 2579–2593. [Google Scholar] [CrossRef]
- Ge, X.; Dietrich, C.; Matsuno, M.; Li, G.; Berg, H.; Xia, Y. An Arabidopsis aspartic protease functions as an anti-cell-death component in reproduction and embryogenesis. EMBO Rep. 2005, 6, 282–288. [Google Scholar] [CrossRef]
- Chen, H.-J.; Huang, Y.-H.; Huang, G.-J.; Huang, S.-S.; Chow, T.-J.; Lin, Y.-H. Sweet potato SPAP1 is a typical aspartic protease and participates in ethephon-mediated leaf senescence. J. Plant Physiol. 2015, 180, 1–17. [Google Scholar] [CrossRef]
- Liu, Y.; Su, M.; Zhao, X.; Liu, M.; Wu, J.; Wu, X.; Lu, Z.; Han, Z. Combined transcriptomic and metabolomic analysis revealed the salt tolerance mechanism of Populus talassica × Populus euphratica. BMC Plant Biol. 2025, 25, 361. [Google Scholar] [CrossRef]
- Wang, J.Y.; Wang, J.P. A Populus euphratica NAC protein regulating Na+/K+ homeostasis improves salt tolerance in Arabidopsis thaliana. Gene 2013, 521, 265–273. [Google Scholar] [CrossRef]
- Li, J.; Zheng, G.; Zhao, J.; Yang, Y.; Meng, H.; Jia, H. A novel Populus euphratica DUB gene, PeMINDY3, enhances drought and salt tolerance by promoting ROS scavenging. Environ. Exp. Bot. 2024, 220, 105686. [Google Scholar] [CrossRef]
- Mustafa, G.; Komatsu, S. Quantitative proteomics reveals the effect of protein glycosylation in soybean root under flooding stress. Front. Plant Sci. 2014, 5, 627. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Figueiredo, L.; Santos, R.B.; Figueiredo, A. Defense and Offense Strategies: The Role of Aspartic Proteases in Plant-Pathogen Interactions. Biology 2021, 10, 75. [Google Scholar] [CrossRef]
- Lim, L.; Senba, H.; Kimura, Y.; Yokota, S.; Doi, M.; Yoshida, K.I.; Takenaka, S. Influences of N-linked glycosylation on the biochemical properties of aspartic protease from Aspergillus glaucus MA0196. Process Biochem. 2019, 79, 74–80. [Google Scholar] [CrossRef]
- Zhang, M.J.; Liu, Y.L.; Han, G.L.; Zhang, Y.; Wang, B.S.; Chen, M. Salt tolerance mechanisms in trees: Research progress. Trees 2021, 35, 717–730. [Google Scholar] [CrossRef]
- Guo, R.; Xu, X.; Carole, B.; Li, X.; Gao, M.; Zheng, Y.; Wang, X. Genome-wide identification, evolutionary and expression analysis of the aspartic protease gene superfamily in grape. BMC Genom. 2013, 14, 554. [Google Scholar] [CrossRef]
- Sixto, H.; Grau, J.; Alba, N.; Alía, R. Response to sodium chloride in different species and clones of genus Populus L. Forestry 2005, 78, 93–104. [Google Scholar] [CrossRef]
- Ma, T.; Wang, J.Y.; Zhou, G.K.; Yue, Z.; Hu, Q.J.; Chen, Y.; Liu, B.B.; Qiu, Q.; Wang, Z.; Zhang, J.; et al. Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 2013, 4, 2797. [Google Scholar] [CrossRef]
- Feng, S.; Li, N.; Chen, H.; Liu, Z.; Li, C.; Zhou, R.; Zhang, Y.; Cao, R.; Ma, X.; Song, X. Large-scale analysis of the ARF and Aux/IAA gene families in 406 horticultural and other plants. Mol. Hortic. 2024, 4, 13. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, P.; Lascoux, M.; Meagher, T.R.; Liu, J.; Vendramin, G.G. Rapidly evolving genes and stress adaptation of two desert poplars, Populus euphratica and P. pruinosa. PLoS ONE 2013, 8, e66370. [Google Scholar] [CrossRef]
- Islam, M.S.; Ghosh, A. Evolution, family expansion, and functional diversification of plant aldehyde dehydrogenases. Gene 2022, 829, 146522. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.C.; Li, B.; Tan, X.; Zhu, C.P.; Wu, T.; Feng, S.Y.; Yang, Q.H.; Shen, S.Q.; Yu, T.; et al. Polyploidy events shaped the expansion of transcription factors in Cucurbitaceae and exploitation of genes for tendril development. Hortic. Plant J. 2022, 8, 562–574. [Google Scholar] [CrossRef]
- Guevara, M.G.; Oliva, C.R.; Huarte, M.; Daleo, G.R. An Aspartic Protease with Antimicrobial Activity is Induced after Infection and Wounding in Intercellular Fluids of Potato Tubers. Eur. J. Plant Pathol. 2002, 108, 131–137. [Google Scholar] [CrossRef]
- Li, X.; Li, C.J.; Shi, L.; Lv, G.F.; Liu, Y.X.; Li, X.; Jia, X.J.; Liu, J.Y.; Chen, Y.Q.; Zhu, L.; et al. Jasmonate signaling pathway confers salt tolerance through a NUCLEAR FACTOR-Y trimeric transcription factor complex in Arabidopsis. Cell Rep. 2024, 43, 113825. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Fernando, F.D.; Raul, D.G.; Gabriela, G.M. Overexpression of Arabidopsis aspartic protease APA1 gene confers drought tolerance. Plant Sci. 2020, 292, 110406. [Google Scholar] [CrossRef]
- Xia, Y.; Suzuki, H.; Borevitz, J.; Blount, J.; Guo, Z.; Patel, K.; Dixon, R.A.; Lamb, C. An extracellular aspartic protease functions in Arabidopsis disease resistance signaling. EMBO J. 2004, 23, 980–988. [Google Scholar] [CrossRef]
- Rodrigo, I.; Vera, P.; Conejero, V. Degradation of tomato pathogenesis-related proteins by an endogenous 37-kDa aspartyl endoproteinase. Eur. J. Biochem. 1989, 184, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Swapnil, P.; Yadav, A.K.; Srivastav, S.; Sharma, N.K.; Srikrishna, S.; Rai, A.K. Biphasic ROS accumulation and programmed cell death in a cyanobacterium exposed to salinity (NaCl and Na2SO4). Algal Res. 2017, 23, 88–95. [Google Scholar] [CrossRef]
- Cao, S.Q.; Wang, C.; Ji, H.H.; Guo, M.J.; Cheng, J.Y.; Cheng, Y.X.; Yang, C.P. Functional Characterisation of the Poplar Atypical Aspartic Protease Gene PtAP66 in Wood Secondary Cell Wall Deposition. Forests 2021, 12, 1002. [Google Scholar] [CrossRef]
- Cao, S.Q.; Guo, M.J.; Cheng, J.Y.; Cheng, H.; Liu, X.M.; Ji, H.H.; Liu, G.J.; Cheng, Y.X.; Yang, C.P. Aspartic proteases modulate programmed cell death and secondary cell wall synthesis during wood formation in poplar. J. Exp. Bot. 2022, 73, 6876–6890. [Google Scholar] [CrossRef]
- Yu, X.; Feng, T. The multifaceted roles of plant aspartic proteases. J. Exp. Bot. 2025, eraf147. [Google Scholar] [CrossRef]
- Zhang, S.H.; Wu, Z.H.; Ma, D.; Zhai, J.T.; Han, X.L.; Jiang, Z.B.; Liu, S.; Xu, J.D.; Jiao, P.P.; Li, Z.J. Chromosome-scale assemblies of the male and female Populus euphratica genomes reveal the molecular basis of sex determination and sexual dimorphism. Commun. Biol. 2022, 5, 1186. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, P.; Huang, L.; Cai, H. Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance. Plants 2025, 14, 1930. https://doi.org/10.3390/plants14131930
He P, Huang L, Cai H. Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance. Plants. 2025; 14(13):1930. https://doi.org/10.3390/plants14131930
Chicago/Turabian StyleHe, Peiyang, Lifan Huang, and Hanyang Cai. 2025. "Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance" Plants 14, no. 13: 1930. https://doi.org/10.3390/plants14131930
APA StyleHe, P., Huang, L., & Cai, H. (2025). Genome-Wide Identification and Expression Analysis of Aspartic proteases in Populus euphratica Reveals Candidates Involved in Salt Tolerance. Plants, 14(13), 1930. https://doi.org/10.3390/plants14131930