

Genetic Analysis of Lodging Resistance in 1892S Based on the T2T Genome: Providing a Genetic Approach for the Improvement of Two-Line Hybrid Rice Varieties
Abstract
1. Introduction
2. Results
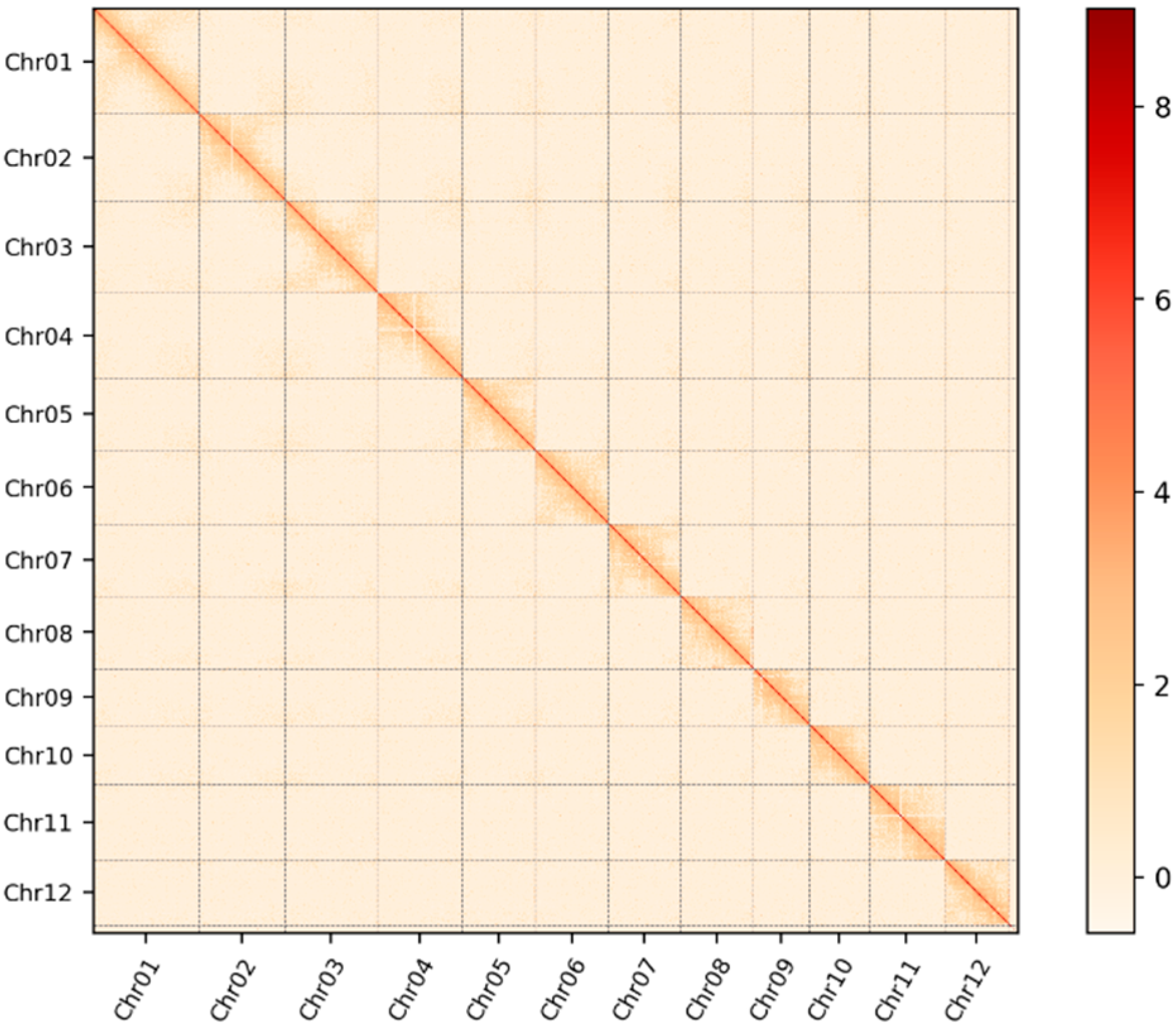
2.1. A Telomere-to-Telomere Gap-Free Genome for 1892S
2.2. T2T Genome Assembly Evaluation of 1892S
2.3. Coding Gene Annotation of 1892S Genome
2.4. Detection of 1892S Lodging Resistance Genes
2.5. Lodging Resistance Evaluation and Validation of 1892S-Based Hybrid Rice Varieties
2.6. Domain-Pan-Genome Dual Screening for Lodging Resistance-Specific Genes
3. Discussion
4. Materials and Methods
4.1. Genomic DNA and RNA Extraction Sequencing
4.2. T2T and Chloroplast Genome Assembly
4.3. Telomere and Centromere Identification
4.4. Genome Assembly Quality Assessment
4.5. Genome Annotation
4.6. Gene Retrieval for Lodging Resistance in Rice
4.7. Genome Comparison Analysis
4.8. Method for Identifying Rice Lodging Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohidem, N.A.; Hashim, N.; Shamsudin, R.; Che Man, H. Rice for Food Security: Revisiting Its Production, Diversity, Rice Milling Process and Nutrient Content. Agriculture 2022, 12, 741. [Google Scholar] [CrossRef]
- Alam, M.; Lou, G.; Abbas, W.; Osti, R.; Ahmad, A.; Bista, S.; Ahiakpa, J.K.; He, Y. Improving Rice Grain Quality Through Ecotype Breeding for Enhancing Food and Nutritional Security in Asia-Pacific Region. Rice 2024, 17, 47. [Google Scholar] [CrossRef]
- Liao, C.; Yan, W.; Chen, Z.; Xie, G.; Deng, X.W.; Tang, X. Innovation and development of the third-generation hybrid rice technology. Crop J. 2021, 9, 693–701. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, J.; Deng, X.W.; Tang, X. Establishment and Advances of Third-Generation Hybrid Rice Technology: A Review. Rice 2023, 16, 56. [Google Scholar] [CrossRef]
- Xu, Y.; Yu, D.; Chen, J.; Duan, M. A review of rice male sterility types and their sterility mechanisms. Heliyon 2023, 9, e18204. [Google Scholar] [CrossRef]
- Zhen, G.; Qin, P.; Liu, K.Y.; Nie, D.Y.; Yang, Y.Z.; Deng, X.W.; He, H. Genome-wide dissection of heterosis for yield traits in two-line hybrid rice populations. Sci. Rep. 2017, 7, 7635. [Google Scholar] [CrossRef]
- Zhang, X.; He, Q.; Zhang, W.; Shu, F.; Wang, W.; He, Z.; Xiong, H.; Peng, J.; Deng, H. Genetic relationships and identification of core germplasm among rice photoperiod- and thermo-sensitive genic male sterile lines. BMC Plant Biol. 2021, 21, 313. [Google Scholar] [CrossRef]
- Li, C.-q.; Pu, Y.-n.; Gao, X.; Cao, Y.; Bao, Y.-y.; Xu, Q.-l.; Du, L.; Tan, J.-r.; Zhu, Y.-h.; Zhang, H.-y.; et al. Detection of quantitative trait nucleotides (QTNs) and QTN-by-environment and QTN-by-QTN interactions for cotton early-maturity traits using the 3VmrMLM method. Ind. Crops Prod. 2024, 216, 118706. [Google Scholar] [CrossRef]
- Wei, X.; Qiu, J.; Yong, K.; Fan, J.; Zhang, Q.; Hua, H.; Liu, J.; Wang, Q.; Olsen, K.M.; Han, B.; et al. A quantitative genomics map of rice provides genetic insights and guides breeding. Nat. Genet. 2021, 53, 243–253. [Google Scholar] [CrossRef]
- Gu, H.; Liang, S.; Zhao, J. Novel Sequencing and Genomic Technologies Revolutionized Rice Genomic Study and Breeding. Agronomy 2022, 12, 218. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, J.; Wang, K.; Han, X.; Yan, T.; Su, Y.; Li, Y.; Lin, Z.; Qin, P.; Fu, C.; et al. The telomere-to-telomere gap-free genome of four rice parents reveals SV and PAV patterns in hybrid rice breeding. Plant Biotechnol. J. 2022, 20, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, L.; Zhang, D. Dissecting the Genetic Basis of Superior Traits in Thermosensitive Genic Male Sterile Line 1892S Through Genome-Wide Analysis. Genet. Mol. Res. 2024, 23, 2024-07. [Google Scholar] [CrossRef]
- Shang, L.; He, W.; Wang, T.; Yang, Y.; Xu, Q.; Zhao, X.; Yang, L.; Zhang, H.; Li, X.; Lv, Y.; et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 2023, 16, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Tong, J.; Han, Z.; Han, A.; Liu, X.; Zhang, S.; Fu, B.; Hu, J.; Su, J.; Li, S.; Wang, S.; et al. Sdt97: A Point Mutation in the 5′ Untranslated Region Confers Semidwarfism in Rice. G3 2016, 6, 1491–1502. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Gui, J.; Fu, C.; Yu, H.; Song, D.; Shen, J.; Qin, P.; Liu, X.; Han, B.; et al. Shortened Basal Internodes Encodes a Gibberellin 2-Oxidase and Contributes to Lodging Resistance in Rice. Mol. Plant 2018, 11, 288–299. [Google Scholar] [CrossRef]
- Rashid, M.A.R.; Zhao, Y.; Azeem, F.; Zhao, Y.; Ahmed, H.G.M.; Atif, R.M.; Pan, Y.; Zhu, X.; Liang, Y.; Zhang, H.; et al. Unveiling the genetic architecture for lodging resistance in rice (Oryza sativa. L) by genome-wide association analyses. Front. Genet. 2022, 13, 960007. [Google Scholar] [CrossRef]
- Ookawa, T.; Hobo, T.; Yano, M.; Murata, K.; Ando, T.; Miura, H.; Asano, K.; Ochiai, Y.; Ikeda, M.; Nishitani, R.; et al. New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat. Commun. 2010, 1, 132. [Google Scholar] [CrossRef]
- Yano, K.; Ookawa, T.; Aya, K.; Ochiai, Y.; Hirasawa, T.; Ebitani, T.; Takarada, T.; Yano, M.; Yamamoto, T.; Fukuoka, S.; et al. Isolation of a novel lodging resistance QTL gene involved in strigolactone signaling and its pyramiding with a QTL gene involved in another mechanism. Mol. Plant 2015, 8, 303–314. [Google Scholar] [CrossRef]
- Yang, X.; Yu, S.; Yan, S.; Wang, H.; Fang, W.; Chen, Y.; Ma, X.; Han, L. Progress in Rice Breeding Based on Genomic Research. Genes 2024, 15, 564. [Google Scholar] [CrossRef]
- Wang, C.; Han, B. Twenty years of rice genomics research: From sequencing and functional genomics to quantitative genomics. Mol. Plant 2022, 15, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.H.; Chen, C.T.; Yang, M.D.; Wu, Y.C.; Lin, C.Y.; Lai, M.H.; Yang, C.Y. Controlling the lodging risk of rice based on a plant height dynamic model. Bot. Stud. 2022, 63, 25. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Koshioka, M.; Kojima, K.; Yoshida, H.; Kawata, M. A role of OsGA20ox1, encoding GA 20-oxidase, in internode elongation of deepwater rice. Plant Physiol. 2004, 135, 1312–1319. [Google Scholar]
- Tong, H.; Jin, Y.; Liu, D.; Liu, L.; Yin, Y.; Qian, Q.; Zhu, L.; Chu, C. DWARF AND LOW-TILLERING, a new member of the GRAS family, plays positive roles in brassinosteroid signaling in rice. G3 2016, 6, 405–413. [Google Scholar] [CrossRef]
- Liu, L.; Tong, H.; Xiao, Y.; Che, R.; Xu, F.; Hu, B.; Liang, C.; Chu, J.; Li, J.; Chu, C. Activation of Big Grain1 significantly improves grain size by regulating auxin transport in rice. PNAS 2018, 112, 11102–11107. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Lu, Y.; Yu, H.; Gu, M.; Liu, Q. The F-box protein APO1 regulates panicle architecture and stem strength in rice. Plant Biotechnol. J. 2021, 19, 620–632. [Google Scholar]
- Jia, L.Q.; Dai, Y.D.; Peng, Z.W.; Cui, Z.B.; Zhang, X.F.; Li, Y.Y.; Tian, W.J.; He, G.H.; Li, Y.; Sang, X.C. The auxin transporter OsAUX1 regulates tillering in rice (Oryza sativa). J. Integr. Agric. 2024, 5, 1454–1467. [Google Scholar] [CrossRef]
- Rashid, M.H.A.; Zhang, X.; Wang, H.; Tang, M.; Liu, X.; Ding, L.; Qian, Q.; Gao, Z. Natural variation in OsFBA2 promotes rice tillering by modulating florigen signaling pathways. Plant Physiol. 2022, 190, 2151–2166. [Google Scholar]
- Ashraf, H.; Ghouri, F.; Baloch, F.S.; Nadeem, M.A.; Fu, X.; Shahid, M.Q. Hybrid Rice Production: A Worldwide Review of Floral Traits and Breeding Technology, with Special Emphasis on China. Plants 2024, 13, 578. [Google Scholar] [CrossRef]
- Cai, Z.; Xu, C.; Liu, X.; Lv, Y.; Ouyang, Y.; Jiang, H. Exploiting male sterility toward the development of hybrid rice. Seed Biol. 2024, 3, e019. [Google Scholar] [CrossRef]
- Cheng, H.; Jarvis, E.D.; Fedrigo, O.; Koepfli, K.P.; Urban, L.; Gemmell, N.J.; Li, H. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 2022, 40, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 2020, 9, giaa094. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Lin, Y.; Ye, C.; Li, X.; Chen, Q.; Wu, Y.; Zhang, F.; Pan, R.; Zhang, S.; Chen, S.; Wang, X.; et al. quarTeT: A telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 2023, 10, uhad127. [Google Scholar] [CrossRef]
- Gao, S.; Yang, X.; Guo, H.; Zhao, X.; Wang, B.; Ye, K. HiCAT: A tool for automatic annotation of centromere structure. Genome Biol. 2023, 24, 58. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- “Picard Toolkit.” Broad Institute, GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 1 May 2025).
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Gabriel, L.; Bruna, T.; Hoff, K.J.; Ebel, M.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 2024, 34, 769–777. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xue, H.; Dong, X.; Li, M.; Zheng, X.; Li, Z.; Xu, J.; Wang, W.; Wei, C. Long-read sequencing of 111 rice genomes reveals significantly larger pan-genomes. Genome Res. 2022, 32, 853–863. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- DB34/T 3924-2021; A Method for Identifying Rice Lodging Resistance. Anhui Provincial Market Supervision and Administration Bureau: Hefei, China, 2021.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomes | Length (bp) | Number of Contigs | Number of Gaps | Centromere Location | Telomere Start Repeat Unit Number | Telomere End Repeat Unit Number |
|---|---|---|---|---|---|---|
| Chr01 | 45,262,897 | 1 | 0 | 16,361,678–19,363,169 | 1404 | 1947 |
| Chr02 | 37,503,140 | 1 | 0 | 13,486,004–15,088,336 | 1786 | 2031 |
| Chr03 | 39,610,054 | 1 | 0 | 20,608,780–21,870,485 | 1607 | 195 |
| Chr04 | 36,637,658 | 1 | 0 | 9,025,928–10,575,559 | 1995 | 1121 |
| Chr05 | 31,267,256 | 1 | 0 | 11,918,420–12,941,910 | 1808 | 804 |
| Chr06 | 31,773,107 | 1 | 0 | 14,604,503–16,500,181 | 904 | 1045 |
| Chr07 | 30,986,196 | 1 | 0 | 11,585,584–14,893,168 | 1042 | 556 |
| Chr08 | 31,327,512 | 1 | 0 | 12,278,862–14,055,598 | 1392 | 1655 |
| Chr09 | 24,231,554 | 1 | 0 | 2,235,737–3,897,225 | 1367 | 877 |
| Chr10 | 25,695,394 | 1 | 0 | 8,608,241–9,522,477 | 1150 | 1550 |
| Chr11 | 32,477,472 | 1 | 0 | 12,858,031–14,900,657 | 1270 | 525 |
| Chr12 | 28,046,869 | 1 | 0 | 9,886,763–11,786,338 | 815 | 1302 |
| Pt | 134,488 | 1 | 0 | - | - | - |
| Gene Name | Gene ID of Nipponbare | Gene ID of 1892S | Reference | Pfams |
|---|---|---|---|---|
| sd1 | Os01g0883800 | Os1892S01G025560 | [14] | DIOX_N, 2OG-FeII_Oxy |
| Sdt97 | Os06g0649800 | Os1892S06G004570 | [15] | Adenine_glyco |
| SBI | LOC_Os05g43880 | Os1892S05G000700 | [16] | DIOX_N, 2OG-FeII_Oxy |
| OsFBA2 | LOC_Os07g09870 | Os1892S07G004930 | [17] | F-box-like, FBA_1 |
| APO1 | Os06g0665400 | Os1892S06G022880 | [18] | F-box |
| OsTB1 | Os03g0706500 | Os1892S03G036300 | [19] | TCP |
| Variety Name | I | II | III | Average Value | LI (Lodging Index) | Level | Phenotype |
|---|---|---|---|---|---|---|---|
| Huiliangyou 27 Zhan | 44.8 | 43.3 | 51.5 | 46.5 | 0.9 | 2 | Moderately strong |
| 21SBC3 | 25.6 | 39.3 | 38.4 | 34.4 | 0.7 | 4 | Very weak |
| 21SBC4 | 59.4 | 55.3 | 51.2 | 55.3 | 1.1 | 2 | Moderately strong |
| 21SBC5 | 55.8 | 52.6 | 58.4 | 55.6 | 1.1 | 2 | Moderately strong |
| 21SBC6 | 39 | 41.4 | 32.3 | 37.6 | 0.7 | 3 | Normal |
| 21SBC7 | 27 | 28.7 | 34.4 | 30.0 | 0.6 | 4 | Very weak |
| 21SBC8 | 25.5 | 30.1 | 22.1 | 25.9 | 0.5 | 4 | Very weak |
| 21SBC9 | 24.4 | 23.6 | 21.2 | 23.1 | 0.5 | 5 | Weak |
| 21SBC10 | 38.8 | 28.8 | 35.9 | 34.5 | 0.7 | 4 | Very weak |
| 21SBC11 | 37.2 | 31.3 | 33.5 | 34.0 | 0.7 | 4 | Very weak |
| 21SBC12 | 26.1 | 36.4 | 35 | 32.5 | 0.6 | 4 | Very weak |
| Huiliangyou 985 | 43.8 | 57.6 | 48.4 | 49.9 | 0.99 | 2 | Moderately strong |
| Wandao 153 (CK) | 49.7 | 49.3 | 51.6 | 50.2 | 1.0 | 2 | Moderately strong |
| Huiliangyou Yuehesimiao | 51.2 | 58.4 | 54.2 | 54.6 | 1.7 | 2 | Moderately strong |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhou, L.; Ni, D.; Ni, J.; Song, F.; Yang, L.; Zhang, D. Genetic Analysis of Lodging Resistance in 1892S Based on the T2T Genome: Providing a Genetic Approach for the Improvement of Two-Line Hybrid Rice Varieties. Plants 2025, 14, 1873. https://doi.org/10.3390/plants14121873
Zhang W, Zhou L, Ni D, Ni J, Song F, Yang L, Zhang D. Genetic Analysis of Lodging Resistance in 1892S Based on the T2T Genome: Providing a Genetic Approach for the Improvement of Two-Line Hybrid Rice Varieties. Plants. 2025; 14(12):1873. https://doi.org/10.3390/plants14121873
Chicago/Turabian StyleZhang, Wei, Liang Zhou, Dahu Ni, Jinlong Ni, Fengshun Song, Liansong Yang, and Dewen Zhang. 2025. "Genetic Analysis of Lodging Resistance in 1892S Based on the T2T Genome: Providing a Genetic Approach for the Improvement of Two-Line Hybrid Rice Varieties" Plants 14, no. 12: 1873. https://doi.org/10.3390/plants14121873
APA StyleZhang, W., Zhou, L., Ni, D., Ni, J., Song, F., Yang, L., & Zhang, D. (2025). Genetic Analysis of Lodging Resistance in 1892S Based on the T2T Genome: Providing a Genetic Approach for the Improvement of Two-Line Hybrid Rice Varieties. Plants, 14(12), 1873. https://doi.org/10.3390/plants14121873