Genome-Wide Identification and Expression Analyses of the MADS-Box Gene During Flowering in Primulina huaijiensis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification and Physicochemical Property Analysis of MADS-Box Gene Family in P. huaijiensis
2.2. Classification and Phylogenetic Analysis of the PhuMADS
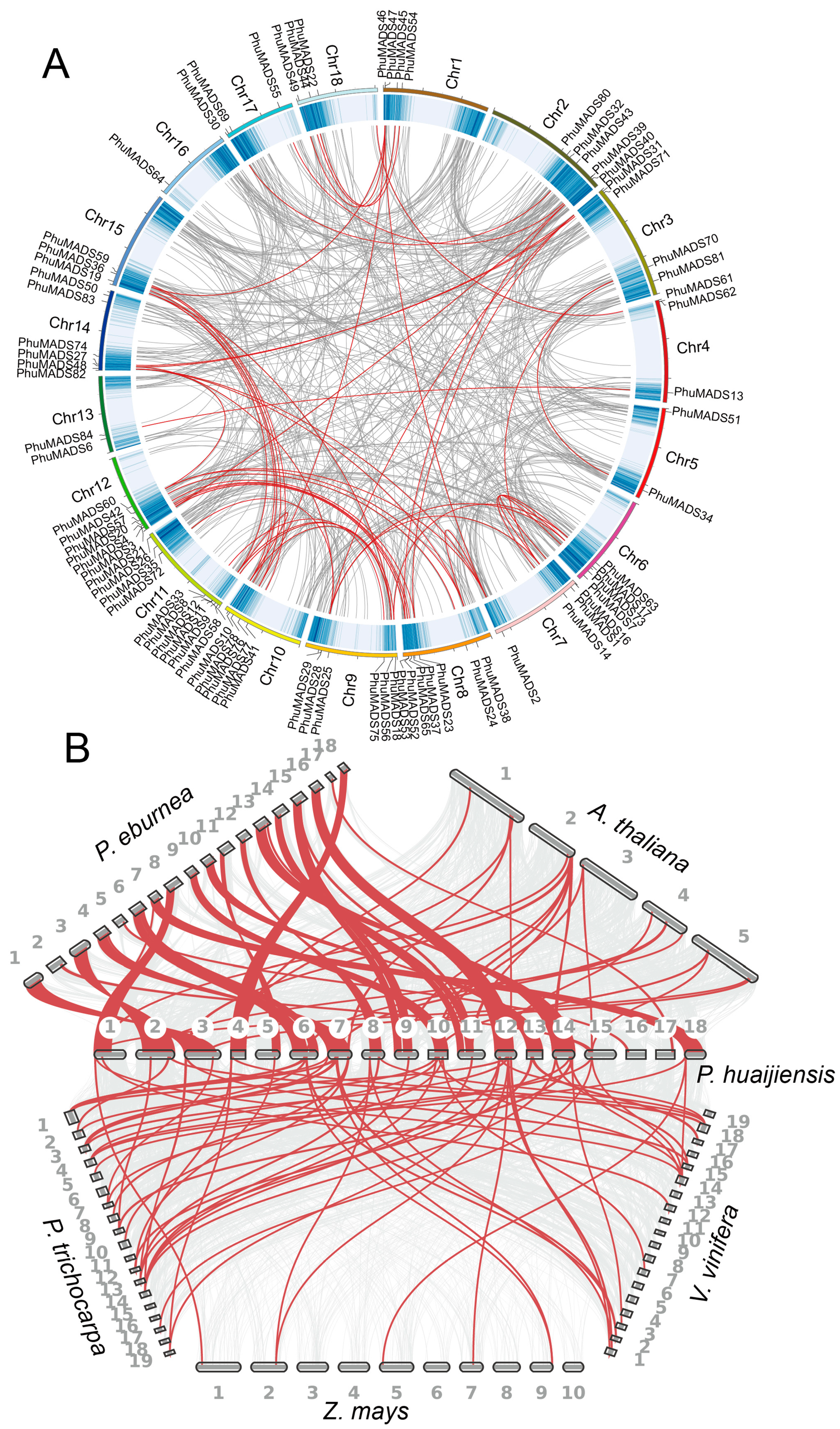
2.3. Chromosomal Localization of the PhuMADS
2.4. Conserved Motif and Gene Structure Analysis of PhuMADS
2.5. Collinearity Analyses of the PhuMADS Within and Between Species
2.6. Cis-Element Analysis of PhuMADS
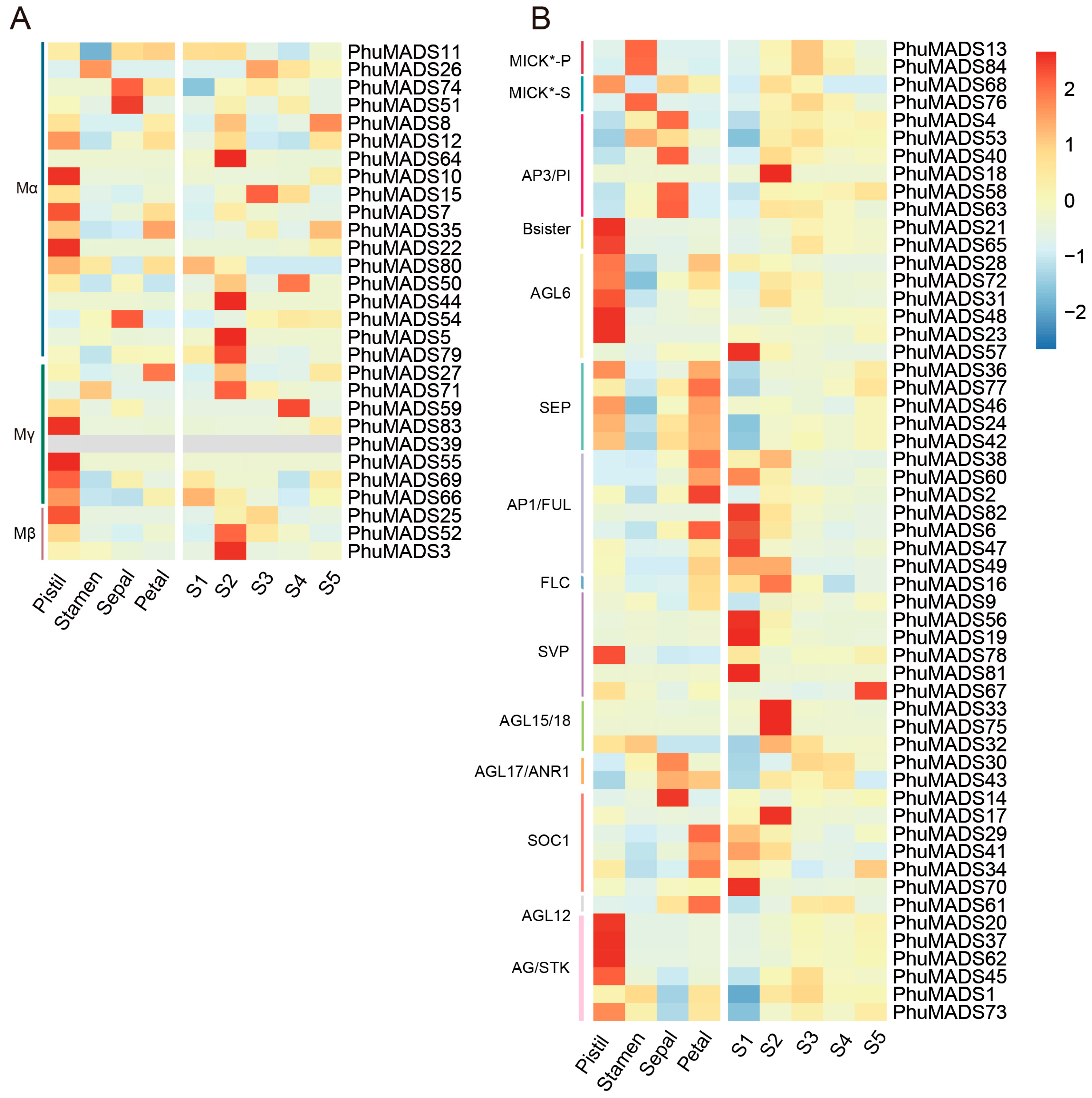
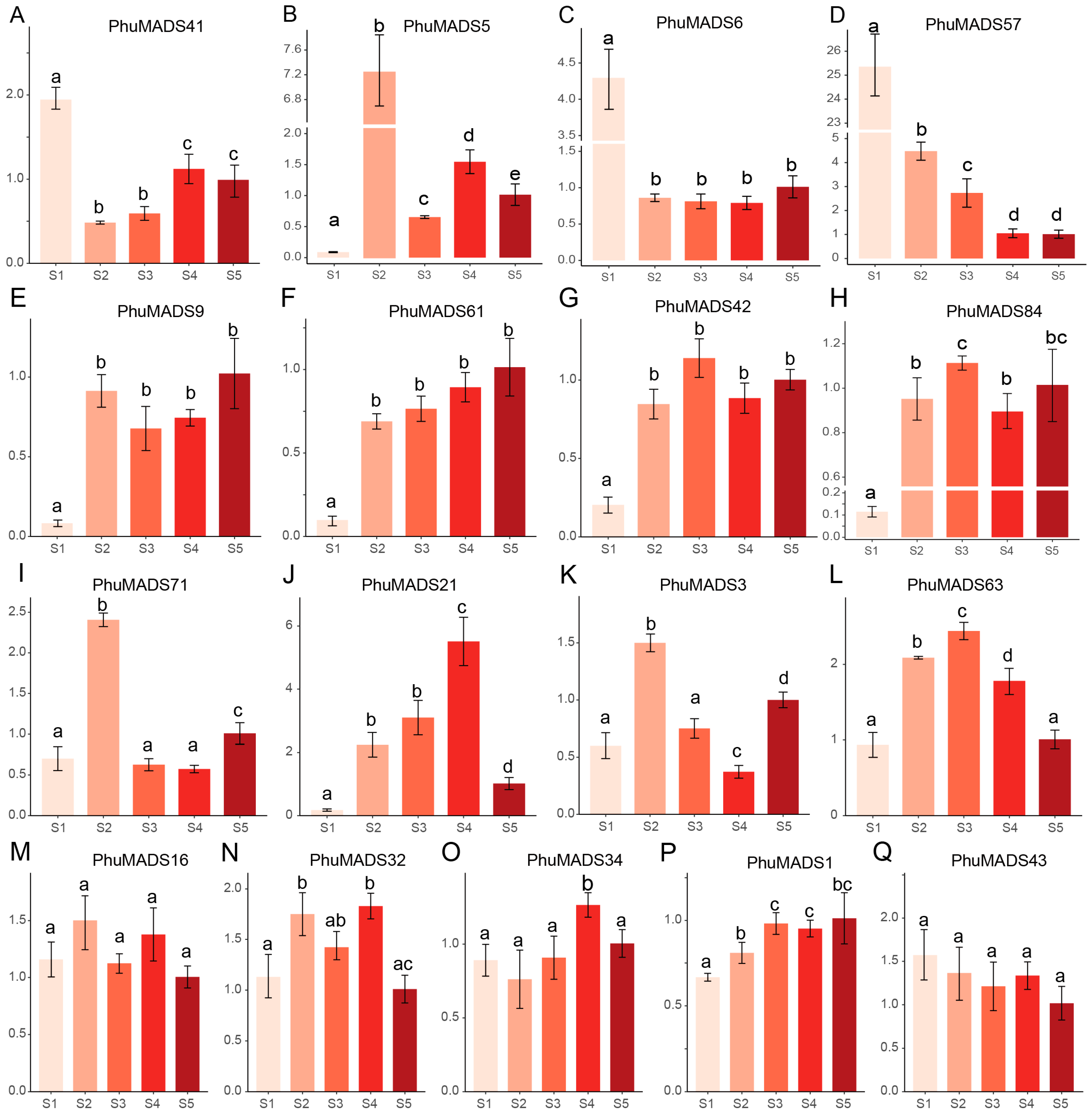
2.7. Expression Profiling of PhuMADS-Box Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Identification of PhuMADS Genes
4.3. Phylogenetic Analysis of PhuMADS Genes
4.4. Chromosomal Location of PhuMADS Genes
4.5. Conserved Motif and Structure Analysis of PhuMADS Genes
4.6. Collinearity Analyses of the PhuMADS Within and Between Species
4.7. Cis-Element Analysis of PhuMADS
4.8. Expression Profiling of PhuMADS
4.9. Expression Analysis of PhuMADS in Different Stage
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alvarez-Buylla, E.R.; Liljegren, S.J.; Pelaz, S.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Vergara-Silva, F.; Yanofsky, M.F. MADS-box gene evolution beyond flowers: Expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2008, 24, 457–466. [Google Scholar]
- Lee, J.H.; Ryu, H.-S.; Chung, K.S.; Posé, D.; Kim, S.; Schmid, M.; Ahn, J.H. Regulation of temperature-responsive flowering by MADS-box transcription factor repressors. Science 2013, 342, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Köhler, C.; de Meaux, J. Endosperm Evolution by Duplicated and Neofunctionalized Type I MADS-Box Transcription Factors. Mol. Biol. Evol. 2022, 39, msab355. [Google Scholar] [CrossRef] [PubMed]
- Passmore, S.; Maine, G.T.; Elble, R.; Christ, C.; Tye, B.-K. Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MATα cells. J. Mol. Biol. 1988, 204, 593–606. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef]
- Sommer, H.; Beltrán, J.; Huijser, P.; Pape, H.; Lönnig, W.; Saedler, H.; Schwarz-Sommer, Z. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: The protein shows homology to transcription factors. EMBO J. 1990, 9, 605–613. [Google Scholar] [CrossRef]
- Gramzow, L.; Ritz, M.S.; Theißen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; de Pouplana, L.R.; Martínez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Münster, T.; Theißen, G. Two Ancient Classes of MIKC-type MADS-box Genes are Present in the Moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Adhikari, P.B.; Kasahara, R.D. An overview on MADS box members in plants: A meta-review. Int. J. Mol. Sci. 2024, 25, 8233. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theißen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef] [PubMed]
- De Bodt, S.; Raes, J.; Van de Peer, Y.; Theißen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003, 8, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fanning, L.; Jack, T. The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 2003, 33, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jack, T. Defining subdomains of the K domain important for protein–protein interactions of plant MADS proteins. Plant Mol. Biol. 2004, 55, 45–59. [Google Scholar] [CrossRef]
- Cho, S.; Jang, S.; Chae, S.; Chung, K.M.; Moon, Y.-H.; An, G.; Jang, S.K. Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol. Biol. 1999, 40, 419–429. [Google Scholar] [CrossRef]
- Lai, X.; Vega-Léon, R.; Hugouvieux, V.; Blanc-Mathieu, R.; van der Wal, F.; Lucas, J.; Silva, C.S.; Jourdain, A.; Muino, J.M.; Nanao, M.H.; et al. The intervening domain is required for DNA-binding and functional identity of plant MADS transcription factors. Nat. Commun. 2021, 12, 4760. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, W.; Lin, P.; Wang, Z.; Chen, Y.; Yang, X.; Zhao, W.; Zhang, Y.; Wang, D.; Que, Y.; et al. Evolution and Function of MADS-Box Transcription Factors in Plants. Int. J. Mol. Sci. 2024, 25, 13278. [Google Scholar] [CrossRef]
- Zik, M.; Irish, V.F. Flower Development: Initiation, Differentiation, and Diversification. Annu. Rev. Cell Dev. Biol. 2003, 19, 119–140. [Google Scholar] [CrossRef]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Huang, B.; Hu, G.; Wang, K.; Frasse, P.; Maza, E.; Djari, A.; Deng, W.; Pirrello, J.; Burlat, V.; Pons, C.; et al. Interaction of two MADS-box genes leads to growth phenotype divergence of all-flesh type of tomatoes. Nat. Commun. 2021, 12, 6892. [Google Scholar] [CrossRef] [PubMed]
- Shah, L.; Sohail, A.; Ahmad, R.; Cheng, S.; Cao, L.; Wu, W. The Roles of MADS-Box Genes from Root Growth to Maturity in Arabidopsis and Rice. Agronomy 2022, 12, 582. [Google Scholar] [CrossRef]
- Ning, Z.L.; Li, G.F.; Wang, J.; Smith, J.F.; Rasolonjatovo, H.; Kang, M. Primulina huaijiensis (Gesneriaceae), a New Species from Guangdong, China. Ann. Bot. Fenn. 2013, 50, 119–122. [Google Scholar] [CrossRef]
- Kang, M.; Tao, J.; Wang, J.; Ren, C.; Qi, Q.; Xiang, Q.; Huang, H. Adaptive and nonadaptive genome size evolution in Karst endemic flora of China. New Phytol. 2014, 202, 1371–1381. [Google Scholar] [CrossRef]
- Morel, P.; Chambrier, P.; Boltz, V.; Chamot, S.; Rozier, F.; Bento, S.R.; Trehin, C.; Monniaux, M.; Zethof, J.; Vandenbussche, M. Divergent Functional Diversification Patterns in the SEP/AGL6/AP1 MADS-Box Transcription Factor Superclade. Plant Cell 2019, 31, 3033–3056. [Google Scholar] [CrossRef]
- Sun, W.; Huang, W.; Li, Z.; Song, C.; Liu, D.; Liu, Y.; Hayward, A.; Liu, Y.; Huang, H.; Wang, Y. Functional and evolutionary analysis of the AP1/SEP/AGL6 superclade of MADS-box genes in the basal eudicot Epimedium sagittatum. Ann. Bot. 2014, 113, 653–668. [Google Scholar] [CrossRef]
- Teotia, S.; Tang, G. To Bloom or Not to Bloom: Role of MicroRNAs in Plant Flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef]
- Pan, X.; Ouyang, Y.; Wei, Y.; Zhang, B.; Wang, J.; Zhang, H. Genome-wide analysis of MADS-box families and their expressions in flower organs development of pineapple (Ananas comosus (L.) Merr.). Front. Plant Sci. 2022, 13, 948587. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Rouse, D.T.; Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. The molecular basis of vernalization: The central role of FLOWERING LOCUS C (FLC). Proc. Natl. Acad. Sci. USA 2000, 97, 3753–3758. [Google Scholar] [CrossRef]
- Wang, P.; Wang, S.; Chen, Y.; Xu, X.; Guang, X.; Zhang, Y. Genome-wide Analysis of the MADS-Box Gene Family in Watermelon. Comput. Biol. Chem. 2019, 80, 341–350. [Google Scholar] [CrossRef]
- Cheng, S.; Jia, M.; Su, L.; Liu, X.; Chu, Q.; He, Z.; Zhou, X.; Lu, W.; Jiang, C. Genome-Wide Identification of the MADS-Box Gene Family during Male and Female Flower Development in Chayote (Sechium edule). Int. J. Mol. Sci. 2023, 24, 6114. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, Z.; Zhu, L.; Gu, Y.; Guo, B.; Lv, C.; Zhu, J.; Xu, R. Genome-wide analysis of the MADS-box gene family involved in salt and waterlogging tolerance in barley (Hordeum vulgare L.). Front. Plant Sci. 2023, 14, 1178065. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef]
- De Bodt, S.; Raes, J.; Florquin, K.; Rombauts, S.; Rouzé, P.; Theißen, G.; Van de Peer, Y. Genome wide structural annotation and evolutionary analysis of the type I MADS-box genes in plants. J. Mol. Evol. 2003, 56, 573–586. [Google Scholar] [CrossRef]
- Zhou, P.; Qu, Y.; Wang, Z.; Huang, B.; Wen, Q.; Xin, Y.; Ni, Z.; Xu, L. Gene Structural Specificity and Expression of MADS-Box Gene Family in Camellia chekiangoleosa. Int. J. Mol. Sci. 2023, 24, 3434. [Google Scholar] [CrossRef]
- Chai, S.; Li, K.; Deng, X.; Wang, L.; Jiang, Y.; Liao, J.; Yang, R.; Zhang, L. Genome-Wide Analysis of the MADS-box Gene Family and Expression Analysis during Anther Development in Salvia miltiorrhiza. Int. J. Mol. Sci. 2023, 24, 10937. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, Z.; Xu, L.; Zhang, L.; Zou, Q. Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships. Genes 2021, 12, 1956. [Google Scholar] [CrossRef]
- Guan, H.; Wang, H.; Huang, J.; Liu, M.; Chen, T.; Shan, X.; Chen, H.; Shen, J. Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination. Plants 2021, 10, 2142. [Google Scholar] [CrossRef]
- Wang, X.; Huang, Q.; Shen, Z.; Baron, G.C.; Li, X.; Lu, X.; Li, Y.; Chen, W.; Xu, L.; Lv, J.; et al. Genome-Wide Identification and Analysis of the MADS-Box Transcription Factor Genes in Blueberry (Vaccinium spp.) and Their Expression Pattern during Fruit Ripening. Plants 2023, 12, 1424. [Google Scholar] [CrossRef]
- Feng, C.; Wang, J.; Wu, L.; Kong, H.; Yang, L.; Feng, C.; Wang, K.; Rausher, M.; Kang, M. The genome of a cave plant, Primulina huaijiensis, provides insights into adaptation to limestone karst habitats. New Phytol. 2020, 227, 1249–1263. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Xiang, C.; Gao, F.; Jakovlić, I.; Lei, H.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, w202–w208. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Zeng, X.; Xu, Z.; Taranto, A.; Lomas, J.S.; Zhang, Y.; Huang, Y.; Wang, Y.; Yim, W.C.; et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta 2024, 3, e211. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Feng, C. Genome-Wide Analysis of MYB Genes in Primulina eburnea (Hance) and Identification of Members in Response to Drought Stress. Int. J. Mol. Sci. 2024, 25, 465. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Cai, X.; Liu, Q.; Lei, Z.; Feng, C. Genome-Wide Identification and Expression Analyses of the MADS-Box Gene During Flowering in Primulina huaijiensis. Plants 2025, 14, 1843. https://doi.org/10.3390/plants14121843
Zhang J, Cai X, Liu Q, Lei Z, Feng C. Genome-Wide Identification and Expression Analyses of the MADS-Box Gene During Flowering in Primulina huaijiensis. Plants. 2025; 14(12):1843. https://doi.org/10.3390/plants14121843
Chicago/Turabian StyleZhang, Jie, Xinxia Cai, Qin Liu, Ziyi Lei, and Chen Feng. 2025. "Genome-Wide Identification and Expression Analyses of the MADS-Box Gene During Flowering in Primulina huaijiensis" Plants 14, no. 12: 1843. https://doi.org/10.3390/plants14121843
APA StyleZhang, J., Cai, X., Liu, Q., Lei, Z., & Feng, C. (2025). Genome-Wide Identification and Expression Analyses of the MADS-Box Gene During Flowering in Primulina huaijiensis. Plants, 14(12), 1843. https://doi.org/10.3390/plants14121843