Root Transcriptome Analysis Identifies Salt-Tolerance Genes in Sweet Corn Chromosome Segment Substitution Lines (CSSLs)

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Differential Seedling Responses to Salt Stress Between D55 and D96
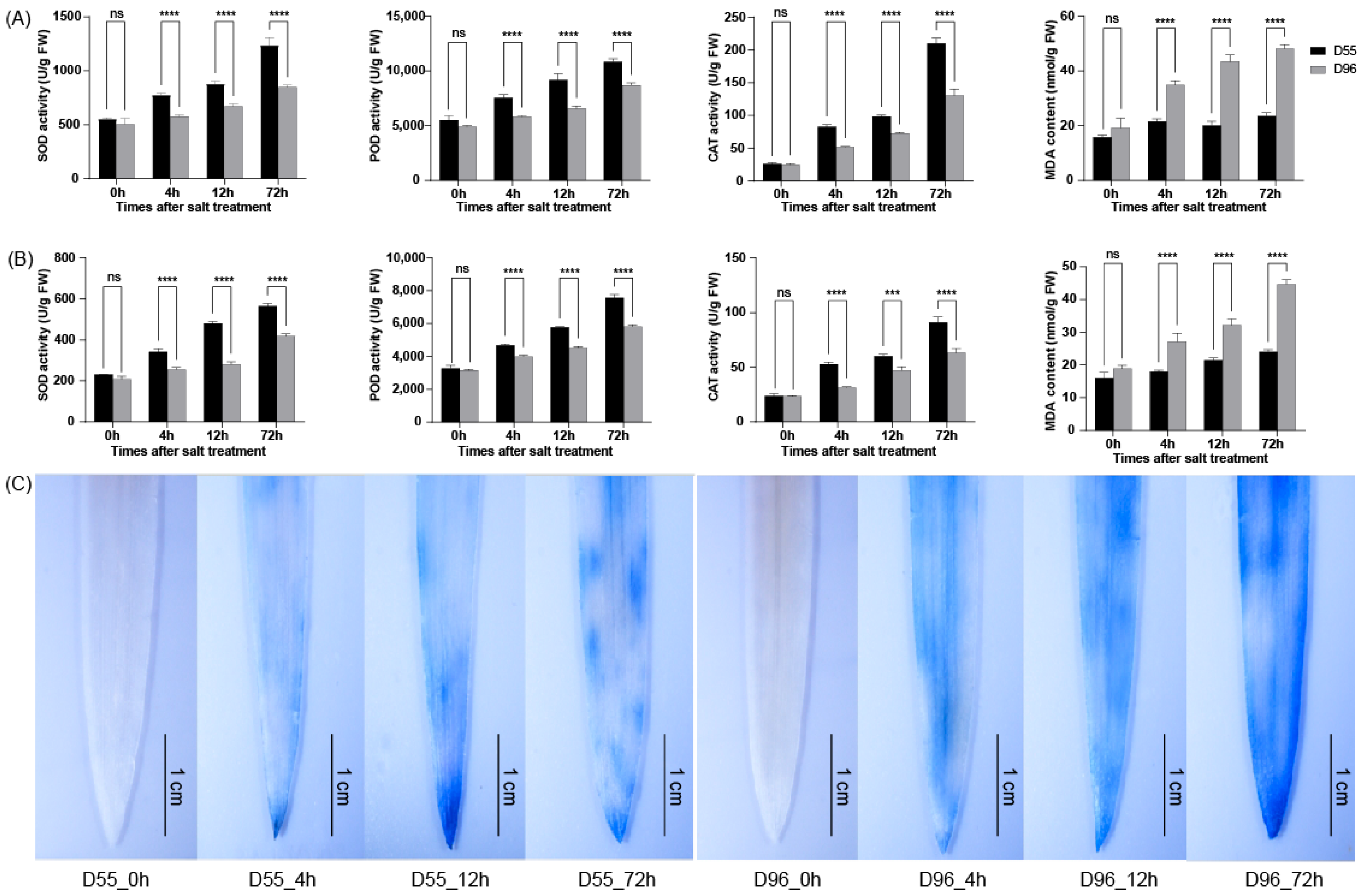
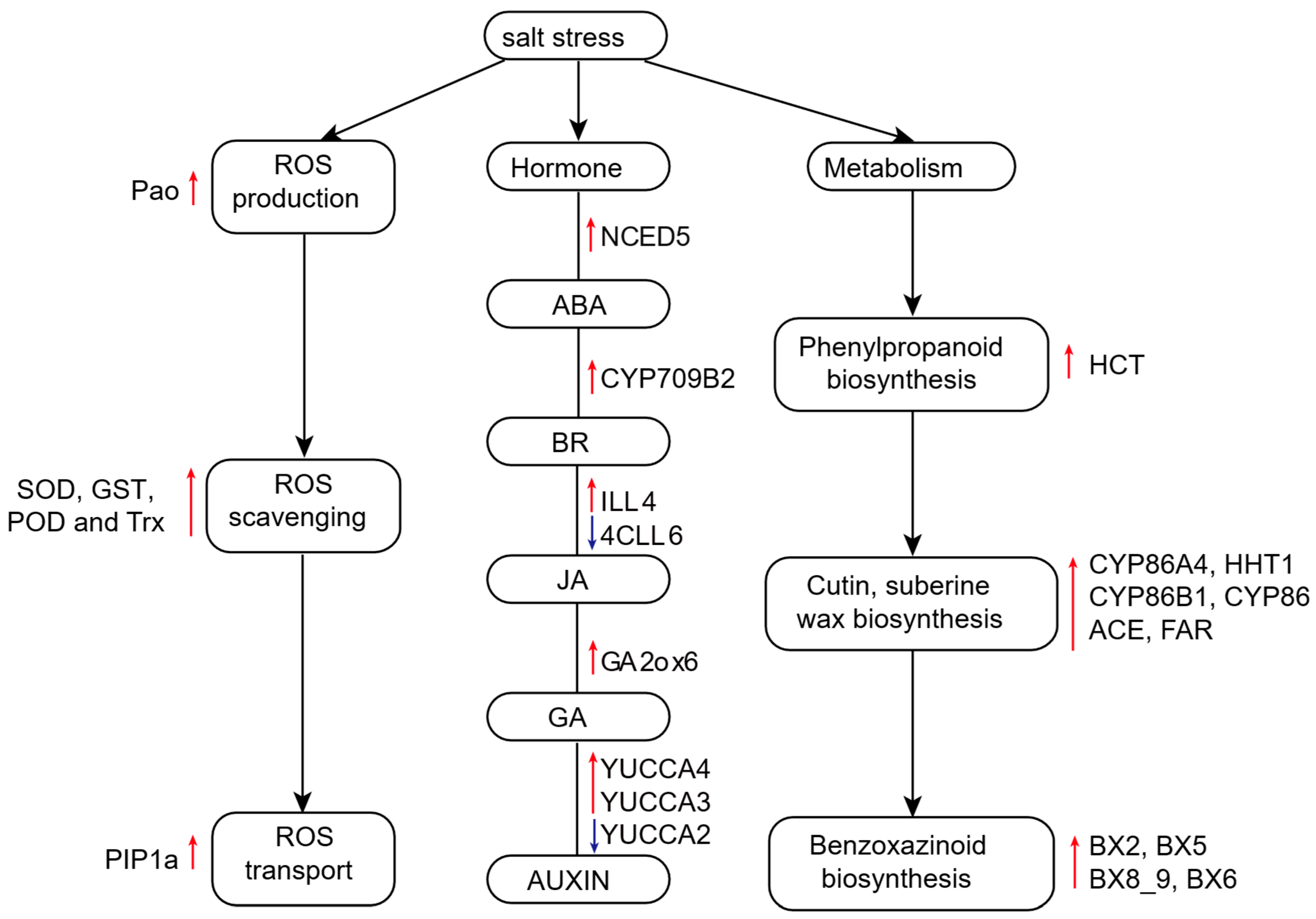
2.2. Enhanced ROS Scavenging Capacity in Salt-Tolerant D55
2.3. Transcriptomic Profiling Under Salt Stress
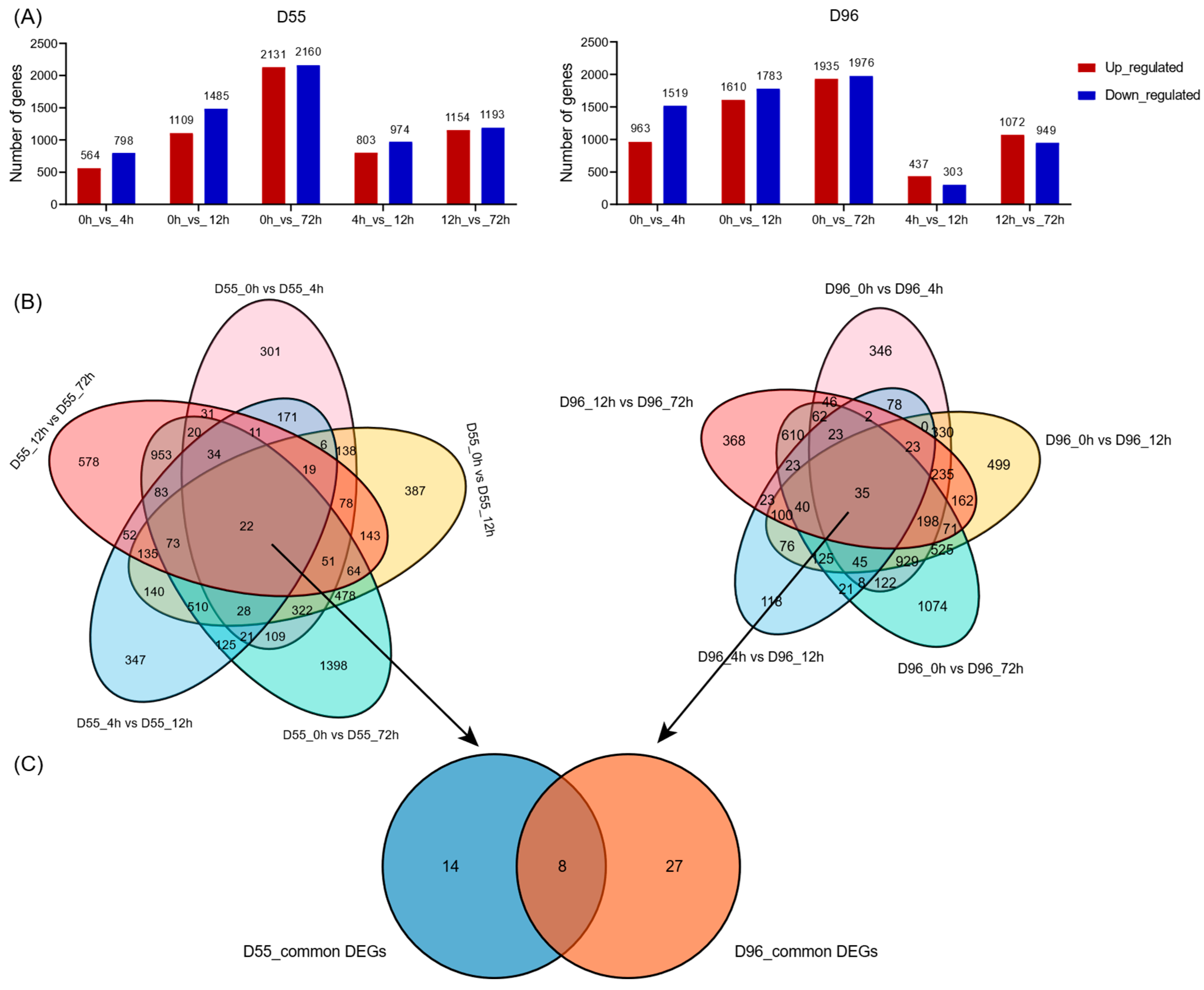
2.4. Dynamic Identification of Salt-Responsive DEGs
2.5. Identification of Core DEGs and Functional Enrichment
2.6. Identification of Salt-Responsive DEGs
2.7. Enhanced ROS Scavenging and Transport Capacity in Salt-Tolerant Genotype D55
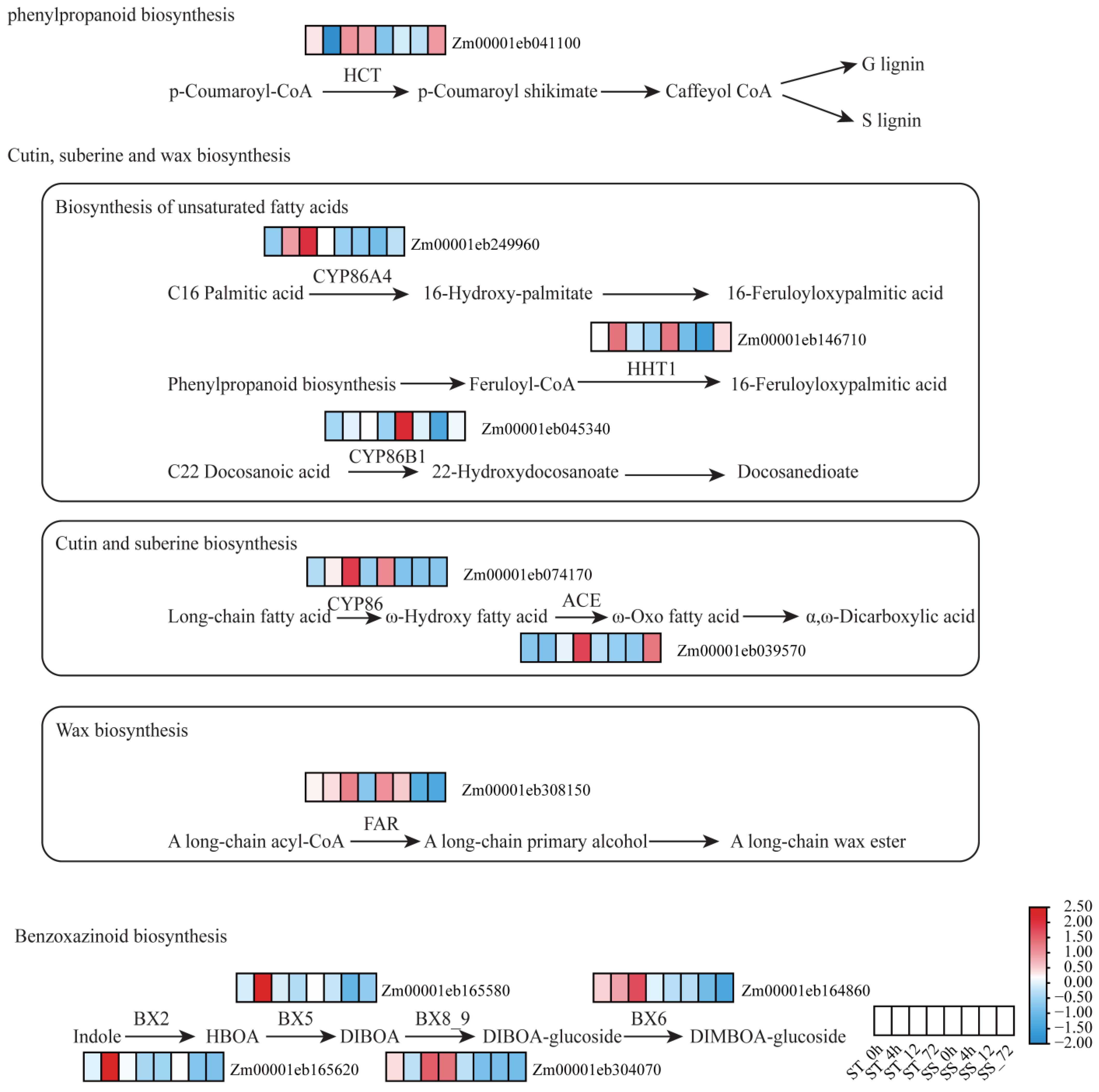
2.8. Upregulation of Phenylpropanoid and Benzoxazinoid Biosynthetic Pathways in D55
2.9. Phytohormone Signaling Divergence Between D55 and D96 Under Salt Stress
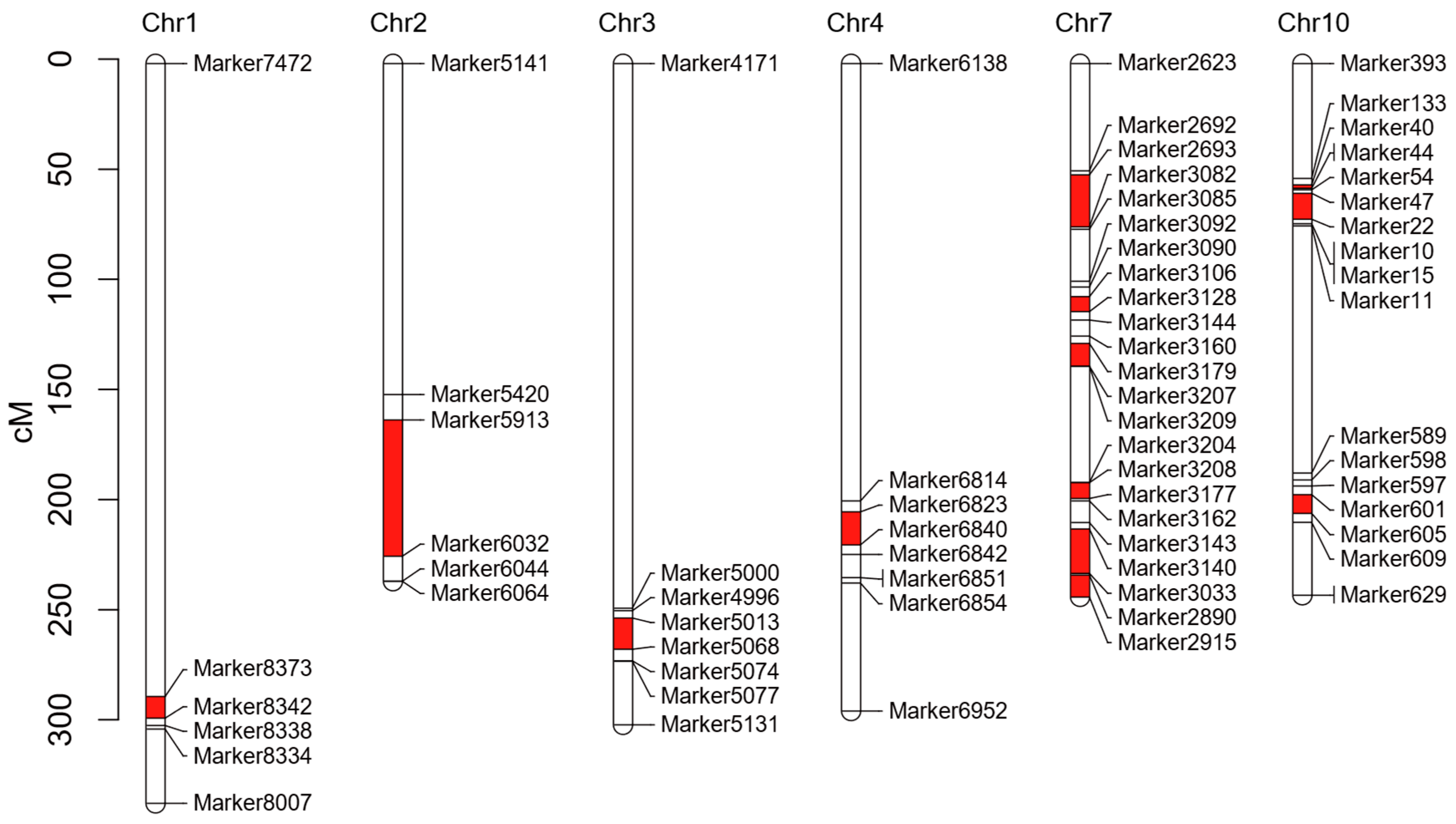
2.10. Transcriptomic and Genetic Variation Analyses Identify Salt Stress-Responsive Candidate Genes
3. Discussion
3.1. Core Persistent DEGs in Salt Stress Response
3.2. ROS Homeostasis Enhancement Underpins Salt Tolerance in D55
3.3. Lignin, Cutin/Suberin/Wax, and Benzoxazine Biosynthesis Promote Salt Tolerance
3.4. Temporal Dynamics of Phytohormone Signaling in Salt Stress Adaptation
4. Materials and Methods
4.1. Plant Materials and Salt Treatment
4.2. Phenotype Observation After Salt Stress
4.3. Antioxidant Activity Determination After Salt Stress
4.4. RNA Extraction, Library Preparation, and Sequencing
4.5. RNA Sequencing Data Analysis
4.6. Quantitative Real-Time PCR
4.7. Combined Analysis of Differential Chromosomal Fragments and Transcriptome
4.8. Variation Analysis of Candidate Genes
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luo, M.; Zhao, Y.; Zhang, R.; Xing, J.; Duan, M.; Li, J.; Wang, N.; Wang, W.; Zhang, S.; Chen, Z.; et al. Mapping of a major QTL for salt tolerance of mature field-grown maize plants based on SNP markers. BMC Plant Biol. 2017, 17, 140. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, B.; Gao, S.; Zhang, F.; Terzaghi, W.; Dai, M. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings. J. Integr. Plant Biol. 2019, 61, 658–674. [Google Scholar] [CrossRef]
- Sandhu, D.; Pudussery, M.V.; Kumar, R.; Pallete, A.; Markley, P.; Bridges, W.C.; Sekhon, R.S. Characterization of natural genetic variation identifies multiple genes involved in salt tolerance in maize. Funct. Integr. Genom. 2020, 20, 261–275. [Google Scholar] [CrossRef]
- Kumar, P.; Choudhary, M.; Halder, T.; Prakash, N.R.; Singh, V.; Vineeth, V.T.; Sheoran, S.; Ravikiran, K.T.; Longmei, N.; Rakshit, S.; et al. Salinity stress tolerance and omics approaches: Revisiting the progress and achievements in major cereal crops. Heredity 2022, 128, 497–518. [Google Scholar] [CrossRef]
- Liang, X.; Li, J.; Yang, Y.; Jiang, C.; Guo, Y. Designing salt stress-resilient crops: Current progress and future challenges. J. Integr. Plant Biol. 2024, 66, 303–329. [Google Scholar] [CrossRef]
- Jha, U.C.; Bohra, A.; Jha, R.; Parida, S.K. Salinity stress response and ‘omics’ approaches for improving salinity stress tolerance in major grain legumes. Plant Cell Rep. 2019, 38, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.J.; Marinova, N.; Lowe, J.A.; Brown, S.; Vellinga, P.; de Gusmao, D.; Hinkel, J.; Tol, R.S. Sea-level rise and its possible impacts given a ‘beyond 4 degrees C world’ in the twenty-first century. Philos. Trans. A Math. Phys. Eng. Sci. 2011, 369, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of Plant Responses to Salt Stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef]
- Wu, H. Plant salt tolerance and Na+ sensing and transport. Crop J. 2018, 6, 215–225. [Google Scholar] [CrossRef]
- Ji, H.; Pardo, J.M.; Batelli, G.; Van Oosten, M.J.; Bressan, R.A.; Li, X. The Salt Overly Sensitive (SOS) pathway: Established and emerging roles. Mol. Plant 2013, 6, 275–286. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, Z.Z.; Zhou, X.F.; Yin, H.B.; Li, X.; Xin, X.F.; Hong, X.H.; Zhu, J.K.; Gong, Z. Overexpression of SOS (Salt Overly Sensitive) genes increases salt tolerance in transgenic Arabidopsis. Mol. Plant 2009, 2, 22–31. [Google Scholar] [CrossRef]
- Ye, C.; Zheng, S.; Jiang, D.; Lu, J.; Huang, Z.; Liu, Z.; Zhou, H.; Zhuang, C.; Li, J. Initiation and Execution of Programmed Cell Death and Regulation of Reactive Oxygen Species in Plants. Int. J. Mol. Sci. 2021, 22, 12942. [Google Scholar] [CrossRef]
- Khazaei, M.; Aghaz, F. Reactive Oxygen Species Generation and Use of Antioxidants during In Vitro Maturation of Oocytes. Int. J. Fertil. Steril. 2017, 11, 63–70. [Google Scholar] [CrossRef]
- Xiao, F.; Zhou, H. Plant salt response: Perception, signaling, and tolerance. Front. Plant Sci. 2022, 13, 1053699. [Google Scholar] [CrossRef] [PubMed]
- Challabathula, D.; Analin, B.; Mohanan, A.; Bakka, K. Differential modulation of photosynthesis, ROS and antioxidant enzyme activities in stress-sensitive and -tolerant rice cultivars during salinity and drought upon restriction of COX and AOX pathways of mitochondrial oxidative electron transport. J. Plant Physiol. 2022, 268, 153583. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Duan, X.; Luo, L.; Dai, S.; Ding, Z.; Xia, G. How Plant Hormones Mediate Salt Stress Responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef]
- Zhang, X.; Han, C.; Liang, Y.; Yang, Y.; Liu, Y.; Cao, Y. Combined full-length transcriptomic and metabolomic analysis reveals the regulatory mechanisms of adaptation to salt stress in asparagus. Front. Plant Sci. 2022, 13, 1050840. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. PPB 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Delgado, C.; Mora-Poblete, F.; Ahmar, S.; Chen, J.T.; Figueroa, C.R. Jasmonates and Plant Salt Stress: Molecular Players, Physiological Effects, and Improving Tolerance by Using Genome-Associated Tools. Int. J. Mol. Sci. 2021, 22, 3082. [Google Scholar] [CrossRef]
- Wang, X.; Yin, J.; Wang, J.; Li, J. Integrative analysis of transcriptome and metabolome revealed the mechanisms by which flavonoids and phytohormones regulated the adaptation of alfalfa roots to NaCl stress. Front. Plant Sci. 2023, 14, 1117868. [Google Scholar] [CrossRef] [PubMed]
- Nidhi; Iqbal, N.; Khan, N.A. Synergistic effects of phytohormones and membrane transporters in plant salt stress mitigation. Plant Physiol. Biochem. PPB 2025, 221, 109685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, P.; Qing, C.; Yang, C.; Shen, Y.; Ma, L. Comparative transcriptome analyses of maize seedling root responses to salt stress. PeerJ 2021, 9, e10765. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, Y.; Liu, H.; Liang, Z.; Zhang, M.; Zou, C.; Yuan, G.; Gao, S.; Pan, G.; Shen, Y.; et al. A Combination of a Genome-Wide Association Study and a Transcriptome Analysis Reveals circRNAs as New Regulators Involved in the Response to Salt Stress in Maize. Int. J. Mol. Sci. 2022, 23, 9755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, B.; Zhang, P.; Han, Q.; Zhao, G.; Zhao, F. Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots. Metabolites 2023, 13, 1155. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Ma, Q.; Zhong, Y.; Zhang, Y.; Zhang, P.; Li, Y.; He, R.; Zhou, Y.; Li, Y.; et al. Integrated single-molecule real-time sequencing and RNA sequencing reveal the molecular mechanisms of salt tolerance in a novel synthesized polyploid genetic bridge between maize and its wild relatives. BMC Genom. 2023, 24, 55. [Google Scholar] [CrossRef]
- Hou, Y.; Zeng, W.; Ao, C.; Huang, J. Integrative analysis of the transcriptome and metabolome reveals Bacillus atrophaeus WZYH01-mediated salt stress mechanism in maize (Zea mays L.). J. Biotechnol. 2024, 383, 39–54. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, Y.; Li, J.; Zhang, P.; Chen, K.; Song, W.; Wang, X.; Yang, J.; Lu, X.; Lu, B.; et al. Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method. Plant Biotechnol. J. 2021, 19, 1937–1951. [Google Scholar] [CrossRef]
- Liang, X.; Liu, S.; Wang, T.; Li, F.; Cheng, J.; Lai, J.; Qin, F.; Li, Z.; Wang, X.; Jiang, C. Metabolomics-driven gene mining and genetic improvement of tolerance to salt-induced osmotic stress in maize. New Phytol. 2021, 230, 2355–2370. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, Y.; Wang, Z.; Wang, Z.Q.; Shi, J.; Liang, X.; Song, W.; Chen, Q.; Lai, J.; Jiang, C. A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na+ exclusion and salt tolerance in maize. New Phytol. 2018, 217, 1161–1176. [Google Scholar] [CrossRef]
- Li, P.; Pan, T.; Wang, H.; Wei, J.; Chen, M.; Hu, X.; Zhao, Y.; Yang, X.; Yin, S.; Xu, Y.; et al. Natural variation of ZmHKT1 affects root morphology in maize at the seedling stage. Planta 2019, 249, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Feng, F.; Zhu, Y.; Xie, F.; Yang, J.; Gong, J.; Liu, Y.; Zhu, W.; Gao, T.; Chen, D.; et al. Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq. Genes 2019, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Brouard, J.S.; Schenkel, F.; Marete, A.; Bissonnette, N. The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J. Anim. Sci. Biotechnol. 2019, 10, 44. [Google Scholar] [CrossRef]
- Ouhaddou, R.; Meddich, A.; Ikan, C.; Lahlali, R.; Ait Barka, E.; Hajirezaei, M.-R.; Duponnois, R.; Baslam, M. Enhancing Maize Productivity and Soil Health under Salt Stress through Physiological Adaptation and Metabolic Regulation Using Indigenous Biostimulants. Plants 2023, 12, 3703. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Liu, H.; Zhang, D.; Hu, M.; Zhang, F.; Ding, S.; Yang, K. Screening of salt tolerance of maize (Zea mays L.) lines using membership function value and GGE biplot analysis. PeerJ 2024, 12, e16838. [Google Scholar] [CrossRef]
- Gibbs, D.J.; Theodoulou, F.L.; Bailey-Serres, J. Primed to persevere: Hypoxia regulation from epigenome to protein accumulation in plants. Plant Physiol. 2024, 197, kiae584. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Li, X.P.; Zhang, T.Q.; Wang, Y.Y.; Wang, C.; Gao, C.Q. Overexpression of ThMYB8 mediates salt stress tolerance by directly activating stress-responsive gene expression. Plant Sci. 2021, 302, 110668. [Google Scholar] [CrossRef]
- Albert, R.; Acharya, B.R.; Jeon, B.W.; Zanudo, J.G.T.; Zhu, M.; Osman, K.; Assmann, S.M. A new discrete dynamic model of ABA-induced stomatal closure predicts key feedback loops. PLoS Biol. 2017, 15, e2003451. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Zhu, D.; Liu, N.; Yan, Y. Endoplasmic Reticulum Subproteome Analysis Reveals Underlying Defense Mechanisms of Wheat Seedling Leaves under Salt Stress. Int. J. Mol. Sci. 2021, 22, 4840. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, C.; Zhang, Q.; Qiu, Z.; Zhang, X.; Hou, Y.; Zang, J. Salinity survival: Molecular mechanisms and adaptive strategies in plants. Front. Plant Sci. 2025, 16, 1527952. [Google Scholar] [CrossRef]
- He, X.; Zhu, J.; Gong, X.; Zhang, D.; Li, Y.; Zhang, X.; Zhao, X.; Zhou, C. Advances in deciphering the mechanisms of salt tolerance in Maize. Plant Signal. Behav. 2025, 20, 2479513. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, Y.Y.; Zhou, Z.C.; Xiang, X.; Liu, X.; Wang, J.; Hu, Z.R.; Xiang, S.P.; Li, W.; Xiao, Q.Z.; et al. Tobacco transcription factor bHLH123 improves salt tolerance by activating NADPH oxidase NtRbohE expression. Plant Physiol. 2021, 186, 1706–1720. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ren, W.; Jiang, S.; Kong, L.; Ma, L.; He, J.; Wang, D.; Liu, W.; Ma, W.; Liu, X. Identification and expression analysis of the RBOH gene family of Isatis indigotica Fort. and the potential regulation mechanism of RBOH gene on H2O2 under salt stress. Plant Cell Rep. 2025, 44, 52. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, L.; Yan, J.; Zhang, J.; Guo, F.; Geng, Y.; Wang, Q.; Yang, F.; Wan, S.; Li, X. Peanut genes encoding tetrapyrrole biosynthetic enzymes, AhHEMA1 and AhFC1, alleviating the salt stress in transgenic tobacco. Plant Physiol. Biochem. PPB 2019, 137, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kesavan, A.K.; Landi, M.; Kaur, S.; Thakur, S.; Zheng, B.; Bhardwaj, R.; Sharma, A. 5-aminolevulinic acid regulates Krebs cycle, antioxidative system and gene expression in Brassica juncea L. to confer tolerance against lead toxicity. J. Biotechnol. 2020, 323, 283–292. [Google Scholar] [CrossRef]
- Zhang, D.J.; Tong, C.L.; Wang, Q.S.; Bie, S. Mycorrhizas Affect Physiological Performance, Antioxidant System, Photosynthesis, Endogenous Hormones, and Water Content in Cotton under Salt Stress. Plants 2024, 13, 805. [Google Scholar] [CrossRef]
- Lu, K.; Chen, X.; Yao, X.; An, Y.; Wang, X.; Qin, L.; Li, X.; Wang, Z.; Liu, S.; Sun, Z.; et al. Phosphorylation of a wheat aquaporin at two sites enhances both plant growth and defense. Mol. Plant 2022, 15, 1772–1789. [Google Scholar] [CrossRef]
- Israel, D.; Lee, S.H.; Robson, T.M.; Zwiazek, J.J. Plasma membrane aquaporins of the PIP1 and PIP2 subfamilies facilitate hydrogen peroxide diffusion into plant roots. BMC Plant Biol. 2022, 22, 566. [Google Scholar] [CrossRef]
- Chun, H.J.; Baek, D.; Cho, H.M.; Lee, S.H.; Jin, B.J.; Yun, D.J.; Hong, Y.S.; Kim, M.C. Lignin biosynthesis genes play critical roles in the adaptation of Arabidopsis plants to high-salt stress. Plant Signal. Behav. 2019, 14, 1625697. [Google Scholar] [CrossRef]
- Tiika, R.J.; Yang, H.; Cui, G.; Ma, Y.; Boamah, S.; Li, Y.; Duan, H. Identification and Analysis of Cuticular Wax Biosynthesis Related Genes in Salicornia europaea Under NaCl Treatment. Int. J. Mol. Sci. 2025, 26, 2632. [Google Scholar] [CrossRef]
- Zhang, C.; Li, J.; Li, S.; Ma, C.; Liu, H.; Wang, L.; Qi, J.; Wu, J. ZmMPK6 and ethylene signalling negatively regulate the accumulation of anti-insect metabolites DIMBOA and DIMBOA-Glc in maize inbred line A188. New Phytol. 2021, 229, 2273–2287. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chi, M.; Liu, S.; Zhang, Y.; Song, J.; Xia, G.; Liu, S. TaGPAT6 enhances salt tolerance in wheat by synthesizing cutin and suberin monomers to form a diffusion barrier. J. Integr. Plant Biol. 2025, 67, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Isayenkov, S.V. The regulation of plant cell wall organisation under salt stress. Front. Plant Sci. 2023, 14, 1118313. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, M.; Ren, T.; Li, K.; Li, Y.; Marowa, P.; Zhang, C. Comparative transcriptome analysis reveals the molecular mechanism of salt tolerance in Apocynum venetum. Plant Physiol. Biochem. PPB 2021, 167, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.K.; Shabala, S. Mechanisms of Plant Responses and Adaptation to Soil Salinity. Innovation 2020, 1, 100017. [Google Scholar] [CrossRef]
- Li, H.; Teng, R.M.; Liu, J.X.; Yang, R.Y.; Yang, Y.Z.; Lin, S.J.; Han, M.H.; Liu, J.Y.; Zhuang, J. Identification and Analysis of Genes Involved in Auxin, Abscisic Acid, Gibberellin, and Brassinosteroid Metabolisms Under Drought Stress in Tender Shoots of Tea Plants. DNA Cell Biol. 2019, 38, 1292–1302. [Google Scholar] [CrossRef]
- Yu, Q.; Cheng, C.; Zhou, X.; Li, Y.; Hu, Y.; Yang, C.; Zhou, Y.; Soliman, T.M.A.; Zhang, H.; Wang, Q.; et al. Ethylene controls cambium stem cell activity via promoting local auxin biosynthesis. New Phytol. 2023, 239, 964–978. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, J.; Wang, W.; Wang, Y.; Xu, J.; Li, Z.; Zhao, X.; Fu, B. Integrated Analysis of the Transcriptome and Metabolome Revealed the Molecular Mechanisms Underlying the Enhanced Salt Tolerance of Rice Due to the Application of Exogenous Melatonin. Front. Plant Sci. 2020, 11, 618680. [Google Scholar] [CrossRef]
- Ma, G.; Zhao, K.; Zhang, Y.; Liu, J.; Chen, M.; Li, X. Dufulin-Binding Protein OsJAZ5 Functions in Rice Stress Tolerance. J. Agric. Food Chem. 2024, 72, 27094–27105. [Google Scholar] [CrossRef]
- Munguia-Rodriguez, A.G.; Lopez-Bucio, J.S.; Ruiz-Herrera, L.F.; Ortiz-Castro, R.; Guevara-Garcia, A.A.; Marsch-Martinez, N.; Carreon-Abud, Y.; Lopez-Bucio, J.; Martinez-Trujillo, M. YUCCA4 overexpression modulates auxin biosynthesis and transport and influences plant growth and development via crosstalk with abscisic acid in Arabidopsis thaliana. Genet. Mol. Biol. 2020, 43, e20190221. [Google Scholar] [CrossRef]
- Shuai, H.; Meng, Y.; Luo, X.; Chen, F.; Zhou, W.; Dai, Y.; Qi, Y.; Du, J.; Yang, F.; Liu, J.; et al. Exogenous auxin represses soybean seed germination through decreasing the gibberellin/abscisic acid (GA/ABA) ratio. Sci. Rep. 2017, 7, 12620. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.; Yoon, Y.; Choi, Y.D. Crosstalk with Jasmonic Acid Integrates Multiple Responses in Plant Development. Int. J. Mol. Sci. 2020, 21, 305. [Google Scholar] [CrossRef]
- Harada, M.; Kubotsu, T.; Agui, T.; Dai, X.; Zhao, Y.; Kasahara, H.; Hayashi, K.I. Investigation of physiological roles of UDP-glycosyltransferase UGT76F2 in auxin homeostasis through the TAA-YUCCA auxin biosynthesis pathway. Biosci. Biotechnol. Biochem. 2024, 88, 1326–1335. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, J.; Xu, X.; Liu, H.; Chang, Q.; Xu, L.; Liang, Z. Genome-Wide Identification of CONSTANS-like (COL) Gene Family and the Potential Function of ApCOL08 Under Salt Stress in Andrographis paniculata. Int. J. Mol. Sci. 2025, 26, 724. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 73. [Google Scholar] [CrossRef]
- Chi, C.; Giri, S.S.; Jun, J.W.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Park, S.C. Detoxification and Immune Transcriptomic Response of the Gill Tissue of Bay Scallop (Argopecten irradians) Following Exposure to the Algicide Palmitoleic Acid. Biomolecules 2018, 8, 139. [Google Scholar] [CrossRef]
- Yang, Y.; Park, S.H.; Alford-Zappala, M.; Lee, H.W.; Li, J.; Cunningham, R.P.; Cao, W. Role of endonuclease III enzymes in uracil repair. Mutat. Res. 2019, 813, 20–30. [Google Scholar] [CrossRef]
- Mu, Z.; Xu, M.; Manda, T.; Yang, L.; Hwarari, D.; Zhu, F.-Y. Genomic survey and evolution analysis of calcium-dependent protein kinases in plants and their stress-responsive patterns in populus. BMC Genom. 2024, 25, 1108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xiao, K.; Li, S.; Li, J.; Huang, J.; Chen, R.; Pang, S.; Zhou, X. Genome-wide analysis of the NAAT, DMAS, TOM, and ENA gene families in maize suggests their roles in mediating iron homeostasis. BMC Plant Biol. 2022, 22, 37. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1–7. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J. TopGO: Enrichment Analysis for Gene Ontology. Bioconductor. Available online: https://www.bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 27 May 2025).
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, C.; Lan, H.; Gao, S.; Liu, H.; Liu, J.; Cao, M.; Pan, G.; Rong, T.; Zhang, S. Validation of potential reference genes for qPCR in maize across abiotic stresses, hormone treatments, and tissue types. PLoS ONE 2014, 9, e95445. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Enzyme | Gene Name | log2FC (D96 vs. D55) | ||
|---|---|---|---|---|---|
| 4 h | 12 h | 72 h | |||
| ROS production | PAO | Zm00001eb070310 | 1.25 | ||
| RBOH | Zm00001eb341910 | −5.56 | −5.53 | −6.67 | |
| Zm00001eb410380 | −1.86 | ||||
| ROS scavenging | SOD | Zm00001eb378880 | 2.06 | 1.88 | 3.06 |
| Zm00001eb226600 | 1.78 | 1.59 | |||
| Zm00001eb420210 | 1.61 | 2.68 | 2.82 | ||
| GST | Zm00001eb141080 | 1.37 | |||
| Zm00001eb002780 | −1.11 | ||||
| Zm00001eb402630 | −1.22 | ||||
| Zm00001eb404560 | −1.97 | −2.98 | −1.87 | ||
| Zm00001eb021720 | −1.37 | ||||
| Zm00001eb021620 | −1.05 | ||||
| POD | Zm00001eb282430 | 1.49 | |||
| Zm00001eb222560 | 5.74 | 6.47 | 9.59 | ||
| Zm00001eb330530 | −1.46 | ||||
| Zm00001eb225230 | −1.15 | −1.04 | |||
| Zm00001eb047120 | 1.23 | ||||
| Trx | Zm00001eb201870 | −2.90 | |||
| ROS transport | PIP | Zm00001eb074210 | 3.49 | 4.40 | 7.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Duan, X.; Liu, P.; Chen, Q.; Sun, W.; Wan, X.; Zheng, Y.; Lin, J.; Jiang, F.; Feng, F. Root Transcriptome Analysis Identifies Salt-Tolerance Genes in Sweet Corn Chromosome Segment Substitution Lines (CSSLs). Plants 2025, 14, 1687. https://doi.org/10.3390/plants14111687
Zhang Z, Duan X, Liu P, Chen Q, Sun W, Wan X, Zheng Y, Lin J, Jiang F, Feng F. Root Transcriptome Analysis Identifies Salt-Tolerance Genes in Sweet Corn Chromosome Segment Substitution Lines (CSSLs). Plants. 2025; 14(11):1687. https://doi.org/10.3390/plants14111687
Chicago/Turabian StyleZhang, Zili, Xuxuan Duan, Pengfei Liu, Qingchun Chen, Wei Sun, Xiaorong Wan, Yixiong Zheng, Jianting Lin, Feng Jiang, and Faqiang Feng. 2025. "Root Transcriptome Analysis Identifies Salt-Tolerance Genes in Sweet Corn Chromosome Segment Substitution Lines (CSSLs)" Plants 14, no. 11: 1687. https://doi.org/10.3390/plants14111687
APA StyleZhang, Z., Duan, X., Liu, P., Chen, Q., Sun, W., Wan, X., Zheng, Y., Lin, J., Jiang, F., & Feng, F. (2025). Root Transcriptome Analysis Identifies Salt-Tolerance Genes in Sweet Corn Chromosome Segment Substitution Lines (CSSLs). Plants, 14(11), 1687. https://doi.org/10.3390/plants14111687